
BP501T – Medicinal Chemistry – Unit-1A (Antihistaminics)

Our Other Brands
Youtube channel

Telegram channel

Table of Contents
Syllabus as per - "PCI"

UNIT- I → Antihistaminics
Study of the development of the following classes of drugs, Classification, mechanism of action, uses of drugs mentioned in the course, Structure activity relationship of selective class of drugs as specified in the course and synthesis ofdrugs superscripted (*)
-
 BP705P – Pharmaceutical Analysis Lab ManualJuly 12, 2024
BP705P – Pharmaceutical Analysis Lab ManualJuly 12, 2024 -
 Medicinal Chemistry-BasicsJuly 3, 2024
Medicinal Chemistry-BasicsJuly 3, 2024
Histamine, receptors and their distribution in the human body
H1–antagonists:
- Diphenhydramine*,
- Dimenhydrinate,
- Doxylamines,
- Clemastine,
- Diphenylphyraline,
- Tripelenamine,
- Chlorcyclizine,
- Meclizine,
- Buclizine,
- Chlorpheniramine,
- Triprolidine*,
- Phenidamine,
- Promethazine*,
- Trimeprazine,
- Cyproheptadine,
- Azatidine,
- Astemizole,
- Loratadine,
- Cetirizine,
- Levocetrazine
- Cromolyn (Mast cell stabilizer)
H2-antagonists:
- Cimetidine*,
- Famotidine,
- Ranitidine.
Gastric Proton pump inhibitors:
- Omeprazole,
- Lansoprazole,
- Rabeprazole,
- Pantoprazole
Histamine and its significance
Introduction-Histamine
Histamine is an organic heterocyclic compound discovered in 1910. It has an imidazole nucleus and is synthesized from histidine, an important amino acid, using histidine decarboxylase. Histamine is produced in the body and stored in basophils and mast cells before being released as an immune response to invading pathogens. In addition to increasing local immunity, it functions as a neurotransmitter in the uterus, brain, and spinal cord. Furthermore, it aids in the regulation of gastrointestinal physiological functions.
Biological role of histamine in human body
Histamine has various important roles in the human body, including in the immunological, digestive, and neurological systems. The following are the key functions and roles of histamine:
Immune Response
Allergic Reactions: Mast cells and basophils produce histamine as part of their immunological response to allergens. This release causes common allergic reactions such as itching, swelling, redness, and increased mucus production.
Inflammation: Histamine promotes inflammation by increasing capillary permeability, allowing white blood cells and proteins to enter afflicted tissues and combat invaders.
Digestive System – For release of gastric acid
Enterochromaffin-like (ECL) cells in the stomach lining release histamine, which stimulates parietal cells to produce gastric acid. This is necessary for proper digestion and nutritional absorption.
Nervous System – as neurotransmitter
Histamine functions as a neurotransmitter in the brain. It is responsible for regulating the sleep-wake cycle, attentiveness, and hunger. Histamine-producing neurons in the hypothalamus play an important role in alertness and cognitive functioning.
Vasodilation
Histamine dilates blood vessels, which can lead to lower blood pressure. This effect is also part of the body’s strategy for enhancing blood flow to areas where the immune system is active.
Smooth Muscle Contraction
In some tissues, histamine produces smooth muscle contraction. For example, in the lungs, histamine can promote bronchoconstriction, which is important in disorders such as asthma.
Chemistry of Histamine

Histamine is made up of an imidazole ring connected to an ethylamine chain, with the amino group of the sidechain protonated under physiological conditions. The imidazole ring of histamine exists in two tautomeric forms in aqueous solution, which are distinguished by which of the two nitrogen atoms is protonated. The ‘tele’ nitrogen is further away from the side chain and is marked by a lowercase tau sign, whereas the ‘pros’ nitrogen is closer to the side chain and is denoted by a pi sign. In solution, the teletautomer, N-H-histamine, is preferred over the prostautomer, N-H-histamine.

Histamine has two basic centres: the aliphatic amino group and any nitrogen atom of the imidazole ring that does not already have a proton. The aliphatic amino group (pKa approximately 9.4) will be protonated under physiological conditions, whereas the second nitrogen of the imidazole ring (pKa 5.8) will not be protonated. As a result, histamine is often protonated to a single-charged cation. Because human blood is slightly basic (typical pH range of 7.35 to 7.45), the primary type of histamine present is monoprotic at the aliphatic nitrogen.
Biosynthesis and Metabolism of Histamine

Diamine oxidase and histamine methyl transferase were shown to be the two main routes for the degradation of histamine. Histamine dosages that are pharmacologically active have a half-life of less than 10 s in rats and 20–30 s in dogs. Histamine levels were initially determined by bioassay in studies, while later methods used fluorometric and radio-enzymatic methods.
Triple response of histamine
Histamine triple reaction contains three components: red spot, flare, and wheal.
- Red spots are caused by capillary vasodilation.
- Flare is a reddening of the surrounding area generated by arteriolar dilation via the axonal reflex.
- Wheal is caused by the expulsion of extracellular fluid from capillaries and venules.
Histamine Receptors
Histamine is released and then acts by binding to its receptor. Histamine receptors come in four different varieties currently: H1, H2, H3, and H4. H1 receptors are found in the peripheral nervous system, H2 receptors are found on the parietal cells of the stomach, H3 receptors are found in the central nervous system, and H4 receptors are found in basophils.
Receptor Type | Class, protein unit and location | Function |
H1 | GPCR – Gq Peripheral and CNS | v Ileum contraction v itching v systemic vasodilatation v broncho-constriction (Allergy-induced asthma) |
H2 | GPCR – Gs Stomach | v Stimulation of gastric acid secretion |
H3 | GPCR – Gi CNS | v Decrease Acetylcholine, Serotonin and Norepinephrine Neurotransmitter release in CNS v Presynaptic auto-receptors |
H4 | GPCR-Gi WBC and basophils | v Mediate mast cell chemotaxis |
Classification
The substances known as antihistaminic agents are those that prevent histamine’s physiological effects. The anti-histaminic agents are classified into –

H1-Receptor Antagonist

Individual Class examples and structure
Antihistaminics → H1-receptor antagonist → First generation → Ethylenediamine derivatives

Antihistaminics → H1-receptor antagonist → First generation → Ethanolamines derivatives

Antihistaminics → H1-receptor antagonist → First generation → Alkylamines derivatives

Antihistaminics → H1-receptor antagonist → First generation → Piperazine derivatives

Antihistaminics → H1-receptor antagonist → First generation → Tricyclic derivatives

Antihistaminics → H1-receptor antagonist → First generation → Tetracyclic derivatives

Antihistaminics → H1-receptor antagonist → Second generation

Antihistaminics → H1-receptor antagonist → Third generation

Antihistaminics → H2-receptor antagonist

Antihistaminics → H3-receptor agonist

Antihistaminics → H4-receptor agonist

Antihistaminics → Mast cell stabilizers

Antihistaminics → Histidine decarboxylase inhibitors

H1-receptor antagonist
General Mode of action of H1-receptor antagonist
The H1 receptor antagonist interacts to the H1 receptor and prevents histamine from attaching to it. Because histamine cannot connect to the receptor, the H1 receptor cannot be activated, which prevents phospholipase C from being activated. There won’t be any release of the secondary messengers 1,4,5-triphosphate (IP3) and (DAG) since phospholipase C has been inactivated. As a result, neither IP3 nor DAG will be able to mobilise calcium, nor will they be able to activate the third messenger protein Kinase C (pKc). Eicosanoid synthesis and release, which are necessary for the creation of autocoids, will eventually cease. As a result, H1 receptor antagonist binds to H1 receptor and prevents the production of autacoids, so suppressing the allergic reaction.

General therapeutic uses of H1-receptor antagonist
H1-receptor antagonists, often known as H1-blockers or antihistamines, are a class of drugs that inhibit histamine from binding to the body’s H1 receptors. These drugs have several therapeutic uses due to their ability to counteract the effects of histamine. Here are some of the most common therapeutic uses for H1 receptor antagonists:
1. Allergy conditions
H1 receptor antagonists are commonly used to treat allergy symptoms such as:
- H1 blockers can alleviate symptoms of seasonal or chronic allergies (hay fever), including sneezing, itching, runny nose, and nasal congestion.
- H1 blockers can reduce itching and hives caused by allergies.
- Allergic conjunctivitis: These medications help alleviate ocular allergies, including redness, itching, and watering.
2. Insomnia
Some H1 blockers, including diphenhydramine, are sedatives. They are occasionally used as sleep aids to promote sleep in those who suffer from intermittent insomnia.
3. Motion sickness
H1-receptor antagonists can alleviate motion sickness symptoms as nausea, vomiting, and dizziness.
4. Pruritus (itching)
H1 blockers can help with itching caused by a range of conditions, including allergic responses, insect bites, and some skin problems.
5. Dermatological disorders
H1 blockers are sometimes used to treat itching and inflammation in dermatological disorders include atopic dermatitis, eczema, and contact dermatitis.
6. Anaphylaxis
In an emergency, H1 blockers may be used to treat anaphylaxis, a severe and potentially deadly allergic reaction. However, they are usually used as an adjuvant treatment in combination with other therapies like epinephrine.
H1-RECEPTOR ANANTGAONIST - Individual Agents
Astemizole
About the drug
It belongs to a family of medications known as second-generation antihistamines, which are intended to be more selective and less sedative than first-generation treatments. Astemizole was initially authorized by the United States Food and Drug Administration (FDA) in 1988, but in 1999, worries about its cardiac adverse effects arose. It was shown to have the potential to cause QT prolongation, a disease that can result in life-threatening abnormal heart beats. As a result, the FDA and other regulatory bodies throughout the world curtailed or discontinued astemizole’s market clearance.
Indication
Astemizole competes with histamine for binding to H1 receptors in the gastrointestinal tract, uterus, major blood vessels, and bronchial muscle. Histaminic activity causes edema, flare, and pruritus, although astemizole’s reversible binding to H1 receptors inhibits these effects. CNS depression is minimal since the medicine does not easily penetrate the blood-brain barrier and largely interacts with H1 receptors in the periphery rather than the brain. Astemizole may also act on H3 receptors, causing negative consequences.
Mechanism of action
Astemizole competes with histamine for binding to H1-receptors in the gastrointestinal system, uterus, major blood arteries, and bronchial muscle. Astemizole’s reversible binding to H1-receptors prevents the production of edema, flare, and pruritus caused by histaminic activity. CNS depression is low because the medication does not readily cross the blood-brain barrier and primarily interacts at H1 receptors in the periphery rather than within the brain. Astemizole may also act on H3-receptors, producing adverse effects.
Metabolism
Almost completely metabolized in the liver and primarily excreted in the feces.

Half-life
1 day
Toxicity
One of the most significant and potentially fatal adverse effects of astemizole is its capacity to lengthen the QT interval on an electrocardiogram (ECG), resulting in torsades de pointes, a kind of cardiac arrhythmia. Astemizole has been linked to a few occurrences of hepatotoxicity (liver damage). While astemizole is less widely used, it can cause CNS symptoms such as drowsiness, dizziness, headache, and fatigue. These impacts can impair cognitive and physical capacities, making tasks like driving or operating machinery dangerous.
Structure

IUPAC Name
1-(4-fluorobenzyl)-N-{1-[2-(4-methoxyphenyl)ethyl]piperidin-4-yl}-1H-benzimidazol-2-amine
Azatidine
About the drug
It is a second-generation antihistamine.
Indication
Azatadine is used to alleviate upper respiratory mucosal congestion in chronic and allergic rhinitis, as well as nasal and eustachian tube congestion.
Mechanism of action
Azatadine appears to compete with histamine for histamine H1 receptors on effector cells. Antihistamines inhibit the pharmacological effects of histamine, which are mediated by H1-receptor activation, lowering the severity of allergic reactions and tissue damage responses involving histamine release.
Metabolism
Hepatic
Toxicity
Overdose symptoms include clumsiness or unsteadiness, seizures, severe drowsiness, flushing or redness of the face, hallucinations, muscle spasms (especially in the neck and back), restlessness, shortness of breath, a shuffling walk, tic-like (jerky) movements of the head and face, trembling and shaking of hands, and insomnia.
Structure

IUPAC Name
4-(6,11-dihydro-5H-benzo [5,6] cyclohepta [1,2-b]pyridin-11-ylidene)-1-methylpiperidine
Buclizine
About the drug
Buclizine is a first-generation antihistamine with antiemetic and anticholinergic effects, classified as a piperazine derivative. Stuart Pharms manufactured it, and the FDA authorized it in 1957.
Indication
To prevent and cure motion sickness-related nausea, vomiting, and dizziness, as well as vertigo (dizziness induced by another medical condition).
Mechanism of action
Vomiting is a protective mechanism that eliminates irritants and other potentially harmful compounds from the upper GI tract. The vomiting center in the medulla of the brain, which contains the chemotrigger zone (CTZ), regulates emesis or vomiting. The vomiting center comprises neurons with muscarinic cholinergic and histamine connections. These neurons are especially critical in relaying signals from the vestibular system to the vomiting center. Motion sickness is mostly induced by overstimulation of these pathways because of a variety of sensory input. As a result, buclizine inhibits histamine receptors in the vomiting center, lowering activity along these pathways. Furthermore, because buclizine is anti-cholinergic, it affects muscarinic receptors.
Metabolism
Hepatic
Structure

IUPAC Name
1-[4-(2-methylpropan-2-yl)benzyl]-4-[(4-chlorophenyl)(phenyl)methyl] piperazine.
Cetirizine
About the drug
Cetirizine, often known as Zyrtec, is a second-generation histamine H1 antagonist used orally. Cetirizine was authorized by the FDA as a prescription-only pharmaceutical in the United States in 1995, and it was later allowed for over-the-counter use in 2007.
Indication
It treats Seasonal Allergic Rhinitis, Chronic Urticaria, and Perennial Allergic Rhinitis.
Mechanism of action
Cetirizine, a hydroxyzine metabolite, is an antihistamine medication. Its primary effects are obtained by selectively inhibiting peripheral H1 receptors.
Metabolism
Cetirizine is largely metabolised by oxidative O-dealkylation producing a metabolite with negligible antihistaminic action. This phase in cetirizine metabolism’s enzyme or enzymes have yet to be discovered.
Half-life
Plasma elimination half-life is 8.3 hours
Toxicity
It has been reported that cetirizine is excreted in human breast milk. Cetirizine should not be used by nursing moms.
Structure

IUPAC Name
2-(2-{4-[(4-chlorophenyl)(phenyl) methyl]piperazin-1-yl}ethoxy)acetic acid
Chlorcyclizine
About the drug
Chlorcyclizine, a piperazine family antihistamine, is a first generation antihistamine. Chlorcyclizine also contains anticholinergic, antiserotonergic, and local anaesthetic effects.
Indication
Chlorcyclizine is generally used for managing the symptoms of allergic rhinitis, also known as hay fever. Chlorcyclizine, in addition to its antihistamine characteristics has been used as an antiemetic (to relieve nausea and vomiting) and as a mild anticholinergic drug to treat symptoms such as motion sickness and vertigo.
Mechanism of action
Chlorcyclizine works by inhibiting the effects of histamine, a substance generated by the body during an allergic reaction. Chlorcyclizine relieves allergic symptoms by inhibiting histamine H1-receptors.
Metabolism
The liver, lungs, kidney, and spleen have high quantities of the N-desmethyl metabolite.
Half-life
about 12 hour
Structure

IUPAC Name
1-[(4-chlorophenyl)(phenyl) methyl]-4-methylpiperazine
Chlorpheniramine
About the drug
One of the most used antihistaminics, it produces less drowsiness and sedation than promethazine. It is also utilized in veterinary medicine.
Indication
To treat-
- rhinitis,
- urticaria,
- allergies,
- the common cold,
- asthma, and
- hay fever.
Mechanism of action
For binding to the histamine H1 receptor, chlorpheniramine competes with histamine. This competition inhibits the activity of endogenous histamine, resulting in temporary relief of histamine-induced allergy symptoms.
Metabolism
Primarily hepatic via Cytochrome P450 (CYP450) enzymes.
Half-life
21-27 hours
Toxicity
Chlorpheniramine, like any medicine, can be hazardous if misused or taken in large quantities. It is critical to adhere to the appropriate dosage and utilize the drug as directed by a healthcare expert or as specified on the product label.
Overdosing on chlorpheniramine can cause a variety of symptoms and side effects, including:
- Depression of the central nervous system: High dosages of chlorpheniramine can cause excessive sedation, drowsiness, confusion, decreased coordination, and even coma.
- Chlorpheniramine has anticholinergic characteristics, meaning it can suppress the function of the neurotransmitter acetylcholine. As a result, you may experience dry mouth, impaired vision, urine retention, constipation, and an elevated heart rate.
- Chlorpheniramine can cause changes in cardiac rhythm, palpitations, and elevated blood pressure at high doses.
- Gastrointestinal effects: Nausea, vomiting, and stomach pain may occur in cases of chlorpheniramine overdose.
Structure

IUPAC Name
N,N-dimethyl-3-(4-chlorophenyl)-3-(pyridin-2-yl)propanamine
Clemastine
About the drug
Clemastine, also known as clemastine fumarate, is a first-generation antihistamine that belongs to the aminoalkyl ether derivative class.
Indication
For relief of allergic rhinitis symptoms such as sneezing, rhinorrhea, pruritus, and acrimation. Also used to treat minor, uncomplicated allergic skin symptoms such as urticaria and angioedema. Used as a self-medication for the temporary relief of typical cold symptoms.
Mechanism of action
Clemastine interacts to the histamine H1 receptor and is a selective histamine H1 antagonist. This inhibits the activity of endogenous histamine, resulting in temporary reduction of histamine-induced allergy symptoms.
Metabolism
The liver mostly metabolizes it by mono- and didemethylation and glucuronide conjugation.
Toxicity
Overdosage effects to antihistamines can range from central nervous system depression to excitement. In youngsters, excitation, hallucinations, ataxia, incoordination, muscle twitching, athetosis, hyperthermia, cyanosis convulsions, tremors, and hyperreflexia prevail initially, followed by postictal depression and cardio-respiratory stoppage. Mild depression may precede convulsions in youngsters. Dry mouth, dilated pupils, flushing of the face, and fever are all frequent symptoms. Adults are more likely to experience CNS depression, which can range from drowsiness to coma.
Structure

IUPAC Name
2-{2-[1-(4-chlorophenyl)-1-phenylethoxy] ethyl}-1-methylpyrrolidine
Cyproheptadine
About the drug
Cyproheptadine is a first-generation antihistamine that is used to treat a variety of medical conditions. It is a tetracyclic antihistamine that inhibits the binding of histamine and serotonin to their respective receptors.
Indication
Cyproheptadine is approved in the United States for the treatment of allergy symptomatologies such as dermatographia, rhinitis, conjunctivitis, and urticaria, as well as adjunctive therapy in the management of anaphylaxis following epinephrine administration. Cyproheptadine is an over-the-counter medication in Canada that is used to relieve pruritus and stimulate appetite. Cyproheptadine is also approved in Australia for the treatment of vascular headaches. The key therapeutic indications of cyproheptadine are-
- Allergic Conditions: Cyproheptadine is commonly used to treat allergic conditions such as allergic rhinitis (hay fever) and allergic conjunctivitis. This effect is produced by inhibition of H1 histamine receptor
- Appetite Stimulation: Cyproheptadine is known for its appetite-stimulating effects and is sometimes prescribed to promote weight gain and stimulate appetite in. It is thought to enhance appetite by antagonizing serotonin receptors in the brain, which can increase food intake.
- Migraine Prevention: Cyproheptadine is also used as a preventive treatment for migraines. It is believed to help prevent migraines by blocking serotonin receptors and reducing the release of inflammatory substances.
- Serotonin Syndrome: Due to its anti-serotonergic effects, it is important to use cyproheptadine with caution. Excessive serotonin receptor blockade can lead to a rare but potentially serious condition called serotonin syndrome, which can cause symptoms such as agitation, confusion, rapid heart rate, high blood pressure, tremors, and fever.
Mechanism of action
Cyproheptadine appears to exert its antihistamine and antiserotonin actions through competing for binding to histamine and serotonin receptors. The ability of cyproheptadine to enhance appetite may be attributed to serotonin antagonism in the hypothalamic appetite centre.
Metabolism
A quaternary ammonium glucuronide compound of cyproheptadine has been discovered as the main metabolite detected in human urine.

Toxicity
Cyproheptadine overdose is likely to result in significant drowsiness (although paradoxical stimulation has been seen in pediatric patients), as well as anticholinergic side effects as dry mouth and flushing.
Structure

IUPAC Name
4-(5H-dibenzo[a,d]cyclohepten-5-ylidene)-1-methyl piperidine
Dimenhydrinate
About the drug
Dimehydrinate was described in the literature for the first time in 1949 and was patented in 1950. Dimenhydrinate is a salt form of Diphenhydramine and 8-chlorotheophylline, with 53%-55.5% dry diphenhydramine and 44%-47% dried 8-chlorotheophylline.
Indication
Dimenhydrinate is used to prevent and cure motion sickness nausea, vomiting, and vertigo.
Mechanism of action
Dimenhydrinate is a theoclate salt that breaks down to form diphenhydramine and 8-chlorotheophylline. While the precise mechanism of action of Dimenhydrinate is uncertain, it is thought to minimize disturbances to equilibrium via anti-muscarinic actions or histamine H1 antagonism. 8-chlorotheophylline may cause excitement by inhibiting adenosine receptors, lowering diphenhydramine-induced sleepiness.
Metabolism
Dimenhydrinate is a theoclate salt that breaks down to form diphenhydramine and 8-chlorotheophylline. UGTs can either N-glucuronidate diphenhydramine to diphenhydramine N-glucuronide or CYP2D6, CYP1A2, CYP2C9, and CYP2C19 can N-demethylate diphenhydramine to N-desmethyldiphenhydramine. The same enzymes can N-demethylate N-desmethyldiphenhydramine again, resulting in N,N-didesmethyldiphenhydramine, which undergoes oxidative deamination to create diphenylmethoxyacetic acid.

Half-life
The plasma elimination half-life of dimenhydrinate is 5-8 hours.
Toxicity
Overdoes in young children can cause hallucinations, seizures, and death. Overdoes in adults can cause sleepiness, seizures, coma, or respiratory depression.
Structure

IUPAC Name
8-chloro-1,3-dimethyl-2,3,6,9-tetrahydro-1H-purine-2,6-dione;
N,N-dimethyl-2-(diphenylmethoxy)ethanamine
Diphenhydramine*
About the drug
Diphenhydramine, better known by its brand name formulation Benadryl, is a first-generation H1 receptor antihistamine that is widely used. It does possess antiemetic, antitussive, hypnotic, and antiparkinsonian effects. Diphenhydramine has also been linked to a variety of neurotransmitter systems that influence behaviour, including as dopamine, norepinephrine, serotonin, acetylcholine, and opioid. As a result, diphenhydramine’s anxiolytic and antidepressant characteristics are currently being investigated.
Indication
Diphenhydramine is a non-prescription, over the counter (OTC) histamine H1 receptor antagonist (H1 antihistamine) used to treat sneezing, runny nose, itchy/watery eyes, itching of nose or throat, insomnia, pruritis, urticaria, insect bites/stings, allergic rashes, and nausea.
Diphenhydramine is useful in adults and paediatric patients (excluding premature infants and neonates) for:
i) allergy relief
ii) active motion sickness treatment; and
iii) use in Parkinson’s disease.
Mechanism of action
Diphenhydramine primarily acts by antagonizing H1 (Histamine 1) receptors. Respiratory smooth muscles, vascular endothelial cells, the gastrointestinal tract (GIT), heart tissue, immunological cells, the uterus, and neurons in the central nervous system (CNS) all have H1 receptors. When the H1 receptor is activated in these tissues, it causes a variety of effects such as increased vascular permeability, vasodilation resulting in flushing, decreased AV node conduction time, stimulation of sensory nerves of the airways resulting in coughing, smooth muscle contraction of the bronchi and the GIT, and eosinophilic chemotaxis, which promotes the allergic immune response.
Ultimately, diphenhydramine acts as an inverse agonist at H1 receptors, reversing the actions of histamine on capillaries and so alleviating allergic response symptoms. Furthermore, as a first-generation antihistamine, diphenhydramine quickly crosses the blood-brain barrier and conversely agonizes the H1 CNS receptors, causing sleepiness and inhibiting the medullary cough center.
H1 receptors are also related to muscarinic receptors. As a result, diphenhydramine also functions as an antimuscarinic. It accomplishes this by acting as a competitive antagonist of muscarinic acetylcholine receptors, which leads to its use as an anti-Parkinson’s medicine.
Finally, diphenhydramine has been shown to be an intracellular sodium channel blocker, suggesting that it may have local anaesthetic characteristics.
Metabolism
Diphenhydramine is rapidly and extensively metabolized in the first pass. In specifically, diphenhydramine is demethylated to N-desmethyldiphenhydramine (the N-desmethyl metabolite), and subsequently this metabolite is demethylated to N,N-didesmethyldiphenhydramine (the N,N-didesmethyl metabolite). The amine moiety of the N,N-didesmethyl metabolite is then used to make acetyl metabolites such as N-acetyl-N-desmethyldiphenhydramine. Additionally, the N,N-didesmethyl metabolite is oxidized to produce the diphenylmethoxyacetic acid metabolite. The remainder of a dose of diphenhydramine taken is excreted unaltered. The metabolites are then conjugated with glycine and glutamine before being eliminated in the urine.
Furthermore, research have revealed that a number of cytochrome P450 isoenzymes, including CYP2D6, CYP1A2, CYP2C9, and CYP2C19, are involved in the N-demethylation that characterizes the major metabolic pathway of diphenhydramine. CYP2D6 has a higher affinity for catalysis with the diphenhydramine substrate than the other isoenzymes discovered. As a result, inducers, or inhibitors of certain CYP enzymes may possibly impact serum concentrations as well as the occurrence and/or severity of adverse effects associated with diphenhydramine intake.

Half-life
The elimination half-life ranges from 2.4-9.3 hours in healthy adults
Toxicity
Overdoes are predicted to produce consequences comparable to those associated with the use of diphenhydramine, such as drowsiness hyperpyrexia, and anticholinergic effects. Overdoes may also cause mydriasis, fever, flushing, anxiety, tremor, dystonic responses, hallucinations, and ECG abnormalities. Rhabdomyolysis, convulsions, delirium, toxic psychosis, arrhythmias, coma, and circulatory collapse may result with a large dosage. Furthermore, at greater doses, especially in youngsters, symptoms of CNS stimulation such as hallucinations and convulsions may occur; at huge levels, coma or circulatory collapse may occur.
Diphenhydramine has been found in breast milk and has been shown to cross the placenta. As a result, this medicine should be taken only when the potential benefit of treatment to the mother overcomes any potential risks to the growing fetus or suckling newborn.
Structure

IUPAC Name
N,N-dimethyl-2-(diphenylmethoxy)ethanamine
Synthesis - Plan A

Synthesis - Plan B

Diphenylpyraline
About the drug
Diphenylpyraline is an alkylamine antihistamine that belongs to the first generation.
Indication
Used to treat allergic rhinitis, hay fever, and allergic skin conditions.
Mechanism of action
Diphenylpyraline, which is used to treat allergies, works by competing with histamine for H1-receptor sites on effector cells. This decreases histamine effects, resulting in temporary relief in allergy symptoms.
Metabolism
Hepatic
Toxicity
Drowsiness, dry mouth, blurred vision, constipation, and urine retention are common diphenylpyraline side effects. These side effects are often moderate and short.
Structure

IUPAC Name
4-(diphenylmethoxy)-1-methyl piperidine
Doxylamine
About the drug
Doxylamine is a first-generation Histamine H1 antagonist with significant sedative properties that belongs to the aminoalkyl ether derivative family.
Indication
Used alone as a short-term sleep aid, and in conjunction with other medications as a nighttime cold and allergy relief medication. In pregnant women, it is also used in conjunction with Vitamin B6 (pyridoxine) to avoid morning sickness.
Mechanism of action
Doxylamine works by blocking histamine at H1 receptors in a competitive manner. It also possesses sedative and anticholinergic properties.
Metabolism
Hepatic
Half-life
10 hours
Toxicity
Overdose symptoms include wheezing, chest tightness, fever, itching, a nasty cough, blue skin tone, fits, and swelling of the face, lips, tongue, or neck.
Structure

IUPAC Name
N,N-dimethyl-2-[1-phenyl-1-(pyridin-2-yl)ethoxy]ethanamine
Levocetrazine
About the drug
Levocetirizine is a histamine H1 antagonist of the third generation. It belongs to the antihistamine diphenylmethylpiperazine class. It is cetirizine’s R enantiomer. Levocetirizine binds to the histamine H1 receptor more strongly than cetirizine. The Belgian pharmaceutical firm introduced levocetirizine in 2001.
Indication
Levocetirizine is used to treat symptoms of chronic allergic rhinitis and simple cutaneous manifestations of chronic idiopathic urticaria. It is also used over the counter to treat a range of minor allergy symptoms.
Mechanism of action
Levocetirizine primarily inhibits histamine H1 receptors. This action inhibits histamine from activating this receptor and causes consequences such as smooth muscle contraction, increased permeability of vascular endothelium, histidine absorption in basophils, activation of cough receptors, and stimulation of flare responses in the nervous system.
Metabolism
Levocetirizine metabolizes slowly, with 85.8% of an oral dosage excreted as the unmodified substance. Levocetirizine may be broken down into dihydrodiol (M2), N-oxide (M3), hydroxymethoxy derivative (M4), hydroxy derivative (M5), O-dealkylated derivative (M6), taurine conjugate (M8), and N-dealkylated and aromatic hydroxylated derivative (M9). M5 metabolite may be glucuronidated to generate M1 metabolite, and M9 metabolite can be glucuronidated to form 4-chloro-4′-hydroxybenzhydryl mercapturates (M10a and M10b).

Half-life
The average half life of levocetirizine is 7.05±1.54 hours
Toxicity
Patients suffering from an overdose may exhibit sleepiness. Before falling asleep, children may get disturbed and restless.
Structure

IUPAC Name
2-(2-{4-[(R)-(4-chlorophenyl)(phenyl) methyl]piperazin-1-yl}ethoxy)acetic acid
Loratadine
About the drug
Loratadine is a tetracyclic derivative antihistamine of the second generation. It is also known as selective histamine H1 receptor antagonists.
Indication
Loratadine is an antihistamine that is used to treat allergic rhinitis, wheal development, urticaria, and other allergic dermatologic disorders.
Mechanism of action
Histamine release has an important role in allergic rhinitis and urticaria. As a result, loratadine works by targeting H1 histamine receptors.
Loratadine binds to H1 histamine receptors on the surface of cells such as epithelial cells, endothelial cells, eosinophils, neutrophils, airway cells, and vascular smooth muscle cells. H1 histamine receptors are classified as G-protein coupled receptors and exist in an equilibrium state between active and inactive variants. Histamine binding to the H1-receptor promotes cross-linking between transmembrane domains III and V, thereby maintaining the receptor’s active form. Antihistamines, on the other hand, bind to a different location on the H1 receptor, favouring the inactive form.
As a result, loratadine is more correctly classed as a “inverse agonist” rather than a “histamine antagonist,” and it can prevent or lessen the severity of histamine-mediated symptoms.
Metabolism
Loratadine is largely metabolized by CYP3A4, CYP2D6, CYP1A1, and CYP2C19 during its first pass metabolism in the liver. CYP1A2, CYP2B6, CYP2C8, CYP2C9, and CYP3A5 are less involved CYP enzymes. The enzymes CYP3A4 and CYP2D6 are primarily responsible for converting loratadine to descarboethoxyloratadine. This major metabolite is four times as potent than loratadine.
Furthermore, a study shows that descarboethoxyloratadine is glucuronidated by UGT2B10 before being hydroxylated by CYP2C8 to create 3-hydroxydesloratadine. The excretion of 3-hydroxydesloratadine is aided by further glucuronidation.
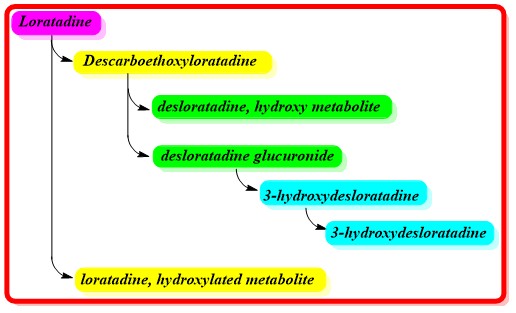
Half-life
Loratadine has an elimination half life of about 10 hours while descarboethoxyloratadine has an elimination half life of about 20 hours.
Toxicity
However, sleeplessness, headache, weariness, sleepiness, and rash have been documented with loratadine. Gastrointestinal side effects, agitation, sleepiness, tachycardia, and headache are all symptoms of loratadine overdose.
Structure

IUPAC Name
Ethyl 4-(8-chloro-5,6-dihydro-11H-benzo[5,6] cyclohepta[1,2-b]pyridin-11-ylidene)-piperidin-1-carboxylate
Meclizine
About the drug
Meclizine is an antihistamine medicine that belongs to the chemical class of piperazine derivatives. It also has anticholinergic effects in addition to being an H1-receptor antagonist.
Indication
Mecilizine is indicated for the symptomatic treatment of motion sickness-related nausea, vomiting, and dizziness, as well as the management of vertigo caused by a variety of conditions such as radiation sickness, Meniere’s syndrome, labyrinthitis, and other vestibular abnormalities.
Mechanism of action
Vomiting is a reflex mechanism that is controlled by the vomiting center and the chemoreceptor trigger zone (CTZ) in the medulla. CTZ also controls motion sickness. The blood-brain barrier at the CTZ is particularly permeable to circulating mediators, and the CTZ can convey impulses to the brainstem vomiting center. Different receptors, including histamine, 5-HT, enkephalins, substance P, and dopamine, are expressed along the brainstem to activate respective pathways and contribute to vomiting regulation.
Histamine H1 receptors are expressed on vestibular nuclei and the nucleus of the solitary tract (NTS), which are triggered by motion sickness and pharyngeal and stomach stimuli. When these nuclei are active, H1 receptor signals is sent to the CTZ and vomiting center.
Meclizine operates primarily by blocking signaling pathway transduction via histaminergic neurotransmission from the vestibular nuclei and NTS to the CTZ and medullary vomiting center via its antagonistic action on the H1 receptors. Meclizine has been shown to reduce labyrinth excitability and vestibular stimulation.
Metabolism
Human evidence on meclizine metabolism is limited. According to in vitro research, meclizine may undergo aromatic hydroxylation or benzylic oxidation via the hepatic CYP2D6 enzyme.

Half-life
In humans, meclizine has a plasma elimination half-life of roughly 5-6 hours.
Toxicity
Overdose symptoms include CNS depression, sleepiness, coma, and convulsions. Hypotension is also possible, especially in the elderly. Anticholinergic effects and CNS stimulation, characterized by hallucinations, convulsions, and difficulty sleeping, are more frequent in children. In the event of an overdose.
Structure

IUPAC Name
1-[(4-chlorophenyl)(phenyl)methyl]-4-[(3-methylphenyl)methyl] piperazine
Phenindamine
About the drug
Phenindamine is a tetracyclic derivative that belongs to the first generation of antihistaminics.
Indication
Sneezing, runny nose, itching, watery eyes, hives, rashes, itching, and other allergy and cold symptoms are treated with phenindamine.
Mechanism of action
On effector cells, phenindamine appears to compete with histamine for histamine H1-receptor sites. Antihistamines block the pharmacological effects of histamine that are mediated by activation of H1-receptor sites, reducing the strength of allergic reactions and tissue damage responses that include histamine release.
Toxicity
Excessive sleepiness, confusion, weakness, ringing in the ears, blurred vision, big pupils, dry mouth, flushing, fever, shaking, sleeplessness, hallucinations, and perhaps seizures are all symptoms of a phenindamine overdose.
Structure

IUPAC Name
2-methyl-9-phenyl-1H,2H,3H,4H,9H-indeno[2,1-c] pyridine
Promethazine*
About the drug
Promethazine, also known as 3,277 R.P., is a phenothiazine N-dimethylaminopropyl derivative discovered in France in 1946.
Indication
Rhinitis, allergic conjunctivitis, allergic reactions to blood or plasma, dermographism, anaphylactic reactions, drowsiness, nausea, vomiting, discomfort, motion sickness, and allergic skin reactions are all treated with promethazine tablets and suppositories. Promethazine cough syrup containing phenylephrine and codeine is used to treat cough, upper respiratory symptoms, and nasal congestion caused by allergies or the common cold.
Mechanism of action
Promethazine is a histamine H1 antagonist, as well as a post-synaptic mesolimbic dopamine, alpha adrenergic, muscarinic, and NMDA receptor antagonist. Antihistamines are used to treat allergic responses. Its anti-muscarinic and anti-NMDA receptor activity contributes to its usage as a sleep aid, as well as for anxiety and tension. Promethazine is effective in the treatment of nausea and vomiting due to its antagonism of histamine H1, muscarinic, and dopamine receptors in the medullary vomiting center.
Metabolism
Promethazine is primarily converted to promethazine sulfoxide, with tiny amounts of desmethylpromethazine and a hydroxy metabolite produced. Promethazine hydroxylation is mostly mediated by CYP2D6.

Half-life
Promethazine has an elimination half life of 12-15 hours.
Toxicity
Promethazine overdose symptoms include mild central nervous system and cardiovascular depression, hypotension, respiratory depression, unconsciousness, hyperreflexia, hypertonia, ataxia, athetosis, extensor-plantar reflexes, convulsions, dry mouth, flushing, gastrointestinal symptoms, and fixed, dilated pupils.
Structure

IUPAC Name
10-[2-(dimethylamino) propyl]-10H-phenothiazine
Synthesis-Plan A

Synthesis-Plan B

Trimeprazine/Alimemazine
About the drug
Trimeprazine is a phenothiazine-class antihistamine medication. Trimeprazine, sometimes known as alimemazine, is a first-generation antihistaminic.
Indication
This medication is used to prevent and treat allergic problems that produce pruritus (itching) and urticaria (certain allergic skin reactions).
Mechanism of action
Trimeprazine competes with free histamine for HA-receptor binding. This inhibits histamine’s effects on HA-receptors, resulting in a reduction in the negative symptoms caused by histamine HA-receptor binding.
Metabolism
Hepatic
Toxicity
Overdose symptoms include clumsiness or unsteadiness, seizures, severe drowsiness, flushing or redness of the face, hallucinations, muscle spasms (particularly in the neck and back), restlessness, shortness of breath, shuffling walk, tic-like (jerky) movements of the head and face, trembling and shaking of the hands, and insomnia.
Structure

IUPAC Name
10-[3-(dimethylamino)-2-methylpropyl]-10H-phenothiazine
Tripelenamine
About the drug
Tripelenamine is an ethlenediamine analogue of the first generation.
Indication
It is used to treat hypersensitivity reactions, coughing, and the common cold.
Mechanism of action
The histamine H1 receptor is bound by tripelennamine. This inhibits the activity of endogenous histamine, resulting in temporary relief of the adverse responses caused by histamine.
Metabolism
Hepatic
Toxicity
Overdoes can cause clumsiness or unsteadiness, convulsions, drowsiness, dryness of the mouth, nose, or throat, feeling faint, flushing or redness of the face, hallucinations, muscle spasms (especially in the neck and back), restlessness, shortness of breath or troubled breathing, shuffling walk, tic-like movements of the head and face, trembling and shaking of hands, and difficulty sleeping.
Structure

IUPAC Name
N-benzyl-N-[(2-dimethylamino)ethyl]pyridin-2-amine
Triprolidine*
About the drug
The antihistaminic triprolidine binds to the H1-receptor. This antihistaminic belongs to the propylamine or alkylamine derivatives class.
Indication
Triprolidine is used to treat the symptoms of seasonal or perennial allergic rhinitis or nonallergic rhinitis, as well as allergic conjunctivitis and moderate, uncomplicated allergic skin manifestations such as urticaria and angioedema. Also used in conjunction with other medicines for the symptomatic reduction of common cold symptoms.
Mechanism of action
Triprolidine binds to the histamine H1 receptor. This inhibits the activity of endogenous histamine, resulting in temporary relief of histamine-induced unpleasant sensations.
Half-life
4 to 6 hours.
Toxicity
Drowsiness, weakness, incoordination, difficulties with micturition, respiratory depression, hypotension, agitation, irritability, convulsions, hypertension, palpitation, and tachycardia are all symptoms of an overdose.
Structure

IUPAC Name
2-[1-(4-methylphenyl)-3-(pyrrolidin-1-yl)prop-1-en-1-yl]pyridine
Synthesis

Structure Activity Relationship of H1-receptor antagonist

At the terminal, histamine has one aromatic ring ethylene bridge and one primary amino functional group. As a result, when developing a histamine antagonist, structural similarity to histamine is required.
To be claimed as a histamine antagonist, additional functional groups must be introduced to the histamine molecule or bioisosteric substitution must be performed. The H1-receptor can recognize a wide range of chemical configurations. As a result, generalized SAR is discussed in greater detail below.
Generalized SAR

The different region of generalized H1 receptor antagonist can be written as-

Instead of a single aryl ring, there are two aryl ring, and instead of a terminal primary amine group, there is a terminal tertiary amine group.
Aryl region
The presence of two aryl rings is required to induce antagonistic properties.

One aryl must be aromatic homocyclic, while the second aryl nucleus can be heterocyclic or aromatic homocyclic.

The presence of an additional substituent on any one aryl preserves the action. Substitution should be added para to the point of attachment, preferably with electron withdrawing groups such as chloro or bromo.
Connecting atom
The connecting atoms are those that exist between the ary ring and the linker. The present H1-receptor blocker contains three types of linking atoms: carba (C), aza (N), and oxa (O). The presence of this linking linker results in a different type of antihistaminic.
The presence of an aza linker results in the formation of ethylenediamine derivatives, the presence of a carba linker results in the creation of alkylamine derivatives, and the presence of a CO linker results in the generation of aminoalkyl ether compounds.

Another class in which the linking atom is aza, but the linker is changed from an aliphatic chain length to a cyclic ring. This belongs to the class of piperazine derivatives.

Linker
- Linker is the part of antihistaminic which connect the connecting atom with terminal amino group.
- In histamine the linker is of 2 carbon chain, in the classical antihistaminics all have two carbon chain linker. This means that any alteration in the length of linker will leads to decrease in activity.
- Addition of small or large functional group on any position of linker will leads to loss in activity.
- Converting the single bond of linker to double bond leads to retain in activity as seen in Triprolidine.

Terminal amino group
In histamine the terminal amino is primary. Converting the primary amino to tertiary leads to produce the compounds that have antagonist activity.

Enclosing the terminal amino in ring form retain the activity only when it possesses the tertiary property.

Increasing the bulk of an aliphatic terminal amino group decreases activity, whereas increasing the bulk of a cyclic tertiary amino group improves activity.

H2-receptor Antagonist
Introduction of H2-receptor antagonist
H2-receptor antagonists, also known as H2 blockers or H2-antihistamines, are a type of medication that prevents histamine from acting on H2 receptors in the stomach. These drugs, which primarily block histamine binding and aim to reduce stomach acid production, are often used for the treatment of diseases such as gastroesophageal reflux disease (GERD), peptic ulcers, and gastritis.
Mode of action of H2-receptor blockers
H2-receptor blockers function by selectively inhibiting histamine activation on H2 receptors in the stomach. Mast cells in the stomach lining release histamine in response to different stimuli, including the presence of food in the stomach. Histamine is produced and binds to H2 receptors on parietal cells. Histamine increases the quantity of the secondary messenger cAMP by activating the H2-receptor. This secondary messenger stimulates the proton pump, causing acid to be poured into the stomach. H2-receptor blockers selectively bind to parietal cell H2 receptors, blocking histamine from binding to these receptors. A decrease in histamine binding results in a decrease in secondary messenger synthesis. When the concentration of secondary messenger is low, the proton pump and acid generation and release are suppressed.
General therapeutic uses of H2-receptor blockers
Because of their potential to lower stomach acid production, H2-receptor blockers have a variety of therapeutic applications. Here are some examples of common therapeutic use for H2-receptor blockers:
1. Gastroesophageal reflux disease (GERD):
H2 blockers are commonly used to treat the symptoms of GERD, a condition in which stomach acid backs up into the esophagus. These medications assist to reduce heartburn, swallowing problems, and other symptoms by reducing acid production.
2. Peptic ulcers
Peptic ulcers are stomach or duodenal lesions that are treated with H2-receptor blockers. By lowering stomach acid levels, H2 blockers improve ulcer healing and prevent recurrence.
3. Gastritis
Gastritis, an inflammation of the stomach lining, is treated with H2 blockers. These medications help to reduce acid production, which alleviates symptoms of gastritis such as stomach discomfort, nausea, and indigestion.
4. Zollinger-Ellison syndrome
H2 blockers are used to treat Zollinger-Ellison syndrome, a rare illness caused by pancreatic or duodenal tumors that causes excessive stomach acid production. By lowering acid release, H2 blockers can reduce elevated acid levels and manage the associated symptoms.
5. Prevention of stress ulcers
H2-receptor blockers can be administered to critically ill patients who are at a higher risk of developing ulcers as a result of factors such as severe illness, trauma, or surgery.
6. Dyspepsia
H2 blockers can help with dyspepsia (indigestion) symptoms like bloating, abdominal discomfort, and early satiety by reducing stomach acid production.
Individual H2 Receptor antagonist
Cimetidine*
About the drug
Cimetidine belongs to the H2-receptor blocker class of medications. It was the first H2 blocker to be released and has been widely utilized for a variety of indications in the past. It suppresses stomach acid secretion as well as the production of pepsin and gastrins. It also inhibits the activity of cytochrome P-450, which may explain why it has been proposed for use in neoadjuvant therapy.
Indications
Cimetidine is used to treat the following conditions: duodenal ulcers, non-malignant gastric ulcers, gastroesophageal reflux disease, and pathological hypersecretion associated with Zollinger-Ellison Syndrome, systemic mastocytosis, and multiple endocrine adenomas. It is used to prevent recurrent gastric or duodenal ulcers, as an adjuvant therapy in the treatment of children with cystic fibrosis, and to treat NSAID-induced lesions and gastrointestinal symptoms.
Mechanism of action
Cimetidine binds to an H2-receptor on the gastric parietal cell’s basolateral membrane, inhibiting histamine effects. This competitive inhibition reduces stomach acid output as well as gastric volume and acidity.
Metabolism
The majority of the parent drug (58-77%) is removed intact in the urine following intravenous injection of cimetidine. Cimetidine sulfoxide is the main metabolite of cimetidine and accounts for around 10-15% of total elimination. A small cimetidine metabolite with a hydroxylated methyl group on the imidazole ring was also discovered, accounting for just 4% of total elimination. Cimetidine metabolism is thought to involve both cytochrome P450 enzymes and flavin-containing monooxygenases, while it is unclear which enzymes are involved.

Half-life
The half-life of cimetidine is predicted to be roughly 2 hours.
Structure

IUPAC name
N”-cyano-N-methyl-N’-(2-{[(5-methyl-1H-imidazol-4-yl)methyl]sulfanyl}ethyl) guanidine
Synthesis – Plan A

Synthesis – Plan B

Famotidine
About the drug
Famotidine is a medicine that belongs to the H2 receptor antagonist class of medications. Famotidine has a high selectivity for the H2 receptor when compared to other H2 receptor antagonists.
Indications
Famotidine is used to treat active duodenal ulcers, active gastric ulcers, symptomatic non-erosive gastroesophageal reflux disease (GERD), and erosive esophagitis caused by GERD in both pediatric and adult patients. It is also used to treat pathological hypersecretory diseases in adults (e.g., Zollinger-Ellison Syndrome, multiple endocrine neoplasias) and to lower the risk of duodenal ulcer recurrence. Famotidine is an over-the-counter medication used to treat and prevent heartburn caused by gastric reflux in children and adults.
Mechanism of action
Histamine functions as a local hormone, stimulating acid production by parietal cells by a paracrine mechanism. Enterochromaffin-like (ECL) cells, which are found near parietal cells, regulate the basal secretion of histamine. Histamine release is also influenced by acetylcholine and gastrin, a peptide hormone. Gastrin is released by Gastrin (G) cells and acts on CCK2 receptors on ECL cells. This action causes ECL cells to produce histamine. Histamine acts on H2 receptors expressed on the basolateral membrane of parietal cells upon release, resulting in elevated intracellular cAMP levels and activated proton pumps. The proton pump pushes additional protons into the stomach, boosting acid secretion.
There is a loss of acid secretion regulation in situations linked with acid hypersecretion, such as ulcers. Famotidine inhibits histamine function by acting on H2 receptors.
Metabolism
Famotidine has a low first-pass metabolism. Hepatic metabolism eliminates about 25-30% of the medication. The S-oxide metabolite is the only one known in humans.

Half-life
The elimination half-life ranges between 2 and 4 hours. In patients with impaired renal function, the half-life is projected to grow non-linearly.
Structure

IUPAC name
3-[({2-[(diaminomethylidene)amino]-1,3-thiazol-4-yl}methyl)sulfanyl]-N’-sulfamoyl propanimidamide
Ranitidine
About the drug
Ranitidine is a medicine that belongs to the H2 receptor antagonist class of medications.
Indications
This medication is used to treat the following disorders: short-term treatment of active duodenal ulcers, treating gastric acid hypersecretion caused by Zollinger-Ellison syndrome, systemic mastocytosis, and other conditions that may pathologically boost gastric acid levels. At a lower dose, it is also utilized in the short-term treatment of active benign gastric ulcers and in the maintenance management of gastric ulcers. In addition to the aforementioned, ranitidine can be used to treat GERD symptoms, endoscopically identified erosive esophagitis, and the maintenance of gastric or duodenal ulcer healing.
Mechanism of action
Following a meal, the hormone gastrin, which is produced by cells in the stomach lining, induces the release of histamine, which subsequently binds to histamine H2 receptors, resulting in the discharge of gastric acid. Ranitidine inhibits stomach acid output by reversibly binding to histamine (H2) receptors on gastric parietal cells. This procedure inhibits histamine binding to this receptor, resulting in a decrease in stomach acid output. Relief of gastric-acid-related symptoms can emerge as soon as 60 minutes after a single dose is administered, and the effects can last 4-10 hours, offering fast and efficient symptomatic relief.
Metabolism
N-oxide is the predominant metabolite in urine, accounting for less than 4% of the dosage. Other ranitidine metabolites include S-oxide (1%), and desmethyl ranitidine (1%). The feces include the remainder of the ranitidine dose excreted.

Half-life
Ranitidine has an elimination half-life of 2.5-3 hours. It could be longer after oral delivery than after injection. The plasma half-life is measured at 3-4 hours for elderly people due to a decline in renal function.
Toxicity
Acute ingestions of up to 18 grams orally were reported to cause transitory unpleasant effects such as tachycardia, bradycardia, dizziness, diarrhea, nausea, and vomiting, among other things. There have also been reports of gait problems and hypotension. When a ranitidine overdose is suspected.
Structure

IUPAC name
N-[1-({2-[({5-[(dimethylamino)methyl]furan-2-yl}methyl)sulfanyl]ethyl}amino)-2-nitroethenyl](methyl)amine
Structure Activity relationship of H2 receptor antagonist
General structure

H2-receptor antagonists are polar chemicals, as opposed to H1-receptor antagonists. The imidazole ring is thought to be critical for H2-receptor antagonist action.
There are two tautomeric forms of the imidazole ring. The variant (I) appears to be necessary for maximal H2-receptor antagonist action.

The discovery of guanylhistamine, which has partial agonist activity at the H2-receptor, is the first step in the evolution of the H2-receptor antagonist.

Burimamide is produced by replacing the quanidino group of guanlyhistamine with thiourea. This drug could not be used orally.

The pKa of the ring is reduced when one of the methyls in the side chain is replaced with an electron withdrawing group such as -S-. A decrease in ring ionization leads to an increase in molecular membrane permeability.
Metiamide is formed by inserting a methyl group into the fourth position of an imidazole ring. This substance can be taken orally.

Cimetidine is produced by replacing the thione (=S) atom of metiamide with cyanoimino (=N-C≡N).

When the imidazole ring of cimetidine is replaced with furan, ranitidine is formed, which is 5 times more powerful than cimetidine.

Famotidine is produced by replacing the furan ring of ranitidine with a thiazole nucleus. Famotidine is 20 times more potent than cimetidine and 7.5 times more effective than ranitidine.

Mast cell stabilizer
Introduction
Mast cells are a type of white blood cell that helps the immune system and the body’s reaction to allergens and infections. They are distributed throughout the body in connective tissues, particularly near blood arteries, nerves, and the skin’s surface.
Mast cells develop from bone marrow progenitor cells and can be found in a variety of tissues, including the skin, respiratory system, and gastrointestinal tract. They include granules carrying bioactive chemicals such as histamine, heparin, cytokines, chemotactic agents, and enzymes such as tryptase and chymase.
Mast cells’ principal role is to serve as immune system sentinels, sensing and responding to foreign chemicals or diseases that enter the body. Mast cells produce granules when triggered, triggering a chain reaction of immunological reactions.
Mast cells play a part in immunological defense against parasites, wound healing, tissue repair, and angiogenesis (the development of new blood vessels) in addition to allergies. They have the ability to interact with other immune cells, such as T-cells and B-cells, and to participate in both innate and adaptive immune responses.
Mast cell stabilizers are a type of drug used to treat and prevent allergic responses that include mast cells. These medications function by inhibiting mast cells from generating histamine and other inflammatory compounds when exposed to allergens or triggers.
When the body has an allergic reaction, mast cells release histamine, leukotrienes, and other substances that induce allergy symptoms such as itching, swelling, redness, and mucus production. Mast cell stabilizers reduce the allergic reaction by preventing the release of these inflammatory chemicals from mast cells.
Mast cell stabilizers work by inhibiting the inflow of calcium ions into mast cells, which is required for the release of histamine and other mediators. Mast cell stabilizers limit mast cell activation and degranulation by blocking calcium influx, hence lowering allergic reaction.
Mast cell stabilizers are frequently used to treat allergies such as hay fever, allergic conjunctivitis (eye allergies), asthma, and atopic dermatitis (eczema). They come in a variety of forms, such as nasal sprays, ocular drops, inhalers, and topical lotions.
Mast cell stabilizers that are commonly administered include cromolyn sodium, nedocromil sodium, and ketotifen. Depending on the formulation and strength, these drugs are available both over-the-counter and via prescription.
Cromoyln sodium/Cromoglicic Acid
About the drug
Cromoglicic acid, often known as cromolyn acid, is the active ingredient in cromolyn sodium. It functions as a mast cell stabilizer. It is used to prevent allergic and exercise-induced asthma attacks, but not to cure existing attacks.
Indications
For the treatment of bronchial asthma patients. Vernal keratoconjunctivitis, vernal conjunctivitis, and vernal keratitis are also treated with this medication.
Mechanism of action
Cromoglicate inhibits mast cell degranulation, preventing the production of histamine and the slow-reacting substance of anaphylaxis (SRS-A), both of which are mediators of type I allergic reactions. Cromoglicate may also inhibit the production of inflammatory leukotrienes. Cromoglicate might work by preventing calcium influx.
Half-life
1.3 hours
Toxicity
Overdose symptoms include coughing, nasal congestion, nausea, sneezing, and wheezing.
Structure

IUPAC name
5-{3-[(2-carboxy-4-oxo-4H-chromen-5-yl)oxy]-2-hydroxypropoxy}-4-oxo- 4H-chromene-2-carboxylic acid
Proton Pump Inhibitor

In parietal cells, the proton pump, a terminal enzyme H+K+ATPase, secretes proton. Acetyl choline activates this terminal enzyme via the M3 Muscarinic receptor, histamine via the H2 receptor, and gastrin via the Gastrin cholecystokinin receptor (CCK2). Prostaglandin is the only proton pump inhibitor, and it inhibits the action of the prostaglandin (EP3) receptor. The activation and inhibition of the proton pump are depicted in the image above.
Proton pump inhibitors are an effective class of medications for treating hyperacidity. This class of medicines binds covalently to the H+K+ATPase enzyme and inhibits it irreversibly. Histamine, gastrin, or acetyl choline will not activate the proton pump via their own receptors.
Proton pump inhibitors establish a covalent disulfide link with the ATPase enzyme, causing the pump to be irreversibly inhibited. One sulfur atom in the disulfide bond will originate from the ATPase’s CYS residue, while the other will come from the proton pump inhibitors. Proton pump inhibitors are particularly effective and long-acting therapeutic agents because the inactivation of the ATPase is irreversible and complete. The ATPase cannot recover from its irreversible interaction with the inhibitor structure, and the body must synthesize a new enzyme from the beginning, which takes time. Gastric acid secretion is delayed until new protein is produced.
Chemistry of Proton Pump
The H+/K+ ATPase proton pump is a big protein that is made up of two subunits: a catalytic alpha subunit and a glycosylated regulatory beta subunit. The alpha subunit has ten transmembrane or membrane-inserted segments and 28 cysteine (CYS) residues.
CYS813 has been identified as the most important residue in the PPIs’ inhibitory activity. This CYS is positioned in the luminal vestibule of the ATPase and is accessible via the ATPase protein’s extracytoplasmic region. Regardless of chemical reactivity, CYS813 will interact with all active PPI structures. PPIs that are triggered more slowly have more time to react with CYS822.
An anionic GLU820 and a second anionic residue (GLU or ASP) at position 824, in addition to CYS813 and CYS822, are thought to be critical in binding the PPI structure to the enzyme and positioning the medication for irreversible interaction with the CYS residue.
Activation of proton pump inhibitors
Proton pump inhibitors are prodrugs, and their activation requires a strongly acidic pH. Proton pump inhibitors are inactive in low acidic or basic pH; hence they must be administered on an empty stomach.
The pH-dependent activation of a proton pump inhibitor is shown in figure below.

Chemistry of Proton Pump Inhibitors
2-pyridylmethylsulfinylbenzimidazole is the proton pump inhibitor pharmacophore. The five current proton pump inhibitor medications (omeprazole, esomeprazole, lansoprazole, pantoprazole, and rabeprazole) all share this basic structural framework and differ mainly in the nature of the substituents added on the pyridine and benzimidazole rings. The structures of omeprazole and esomeprazole are identical. Omeprazole is the racemic form, whereas Esomeprazole is the S-isomer.
The proton pump inhibitor activation pathway starts with two protonation events that happen easily in the extremely acidic parietal cell. Only two of the three nitrogen atoms in the proton pump inhibitor pharmacophore are capable of absorbing proton: the pyridine nitrogen and the doubly linked benzimidazole nitrogen (N3).
Pka1 stood for pyridine nitrogen. Lansoprazole and pantoprazole have a pKa1 of 3.83, Omeprazole and esomeprazole have a pKa1 of 4.06, and Rabeprazole has a pKa1 of 4.53. These pKa1 levels ensure that the pyridine nitrogen of all PPIs is virtually entirely cationic at the parietal cells’ low pH (1.3), keeping the drug directly at the site of action.
The pKa value of benzimidazole N3 (known as pKa2) is significantly lower than that of pyridine nitrogen, ranging from 0.11 (pantoprazole) to 0.79 (omeprazole and esomeprazole). Lansoprazole and rabeprazole have the same pKa2 of 0.62. Because of the lower pKa values, the benzimidazole ring protonates after the pyridine ring, and the extent of protonation is greatly reduced. However, the greater the pKa2 value, the more eagerly the benzimidazole nitrogen takes proton and becomes cationic.
Proton pump inhibitors Individual agents
Omeprazole
About the drug
Omeprazole belongs to the class of medicines known as proton pump inhibitors (PPIs). This was the first clinically useful medicine in its class, and the FDA approved it in 1989.
Indications
Omeprazole, according to the FDA label, is a proton pump inhibitor (PPI) indicated for the following purposes:
- Treatment of active duodenal ulcers in adults
- Helicobacter pylori eradication to lower the incidence of recurrence of duodenal ulcers in adults.
- Treatment of active benign gastric ulcers in adults
- Lowering the risk of upper gastrointestinal (GI) bleeding in critically unwell adult patients.
- Treatment of symptomatic gastroesophageal reflux disease (GERD) in patients 1 year of age and older.
- Treatment of erosive esophagitis due to acid-mediated GERD in patients 1 month of age and older
- Maintenance of erosive esophagitis healing due to acid-mediated GERD in patients 1 year and older
- Pathologic hypersecretory disorders in adults
Mechanism of action
The secretion of hydrochloric acid (HCl) into the gastric lumen is primarily regulated by the proton pump’s H(+)/K(+)-ATPase, which is abundant in the stomach’s parietal cells. ATPase is an enzyme found on the parietal cell membrane that enables hydrogen and potassium exchange, which generally results in potassium extrusion and the creation of HCl (gastric acid).
Omeprazole belongs to a class of antisecretory drugs known as substituted benzimidazoles, which decrease gastric acid secretion by selectively inhibiting the H+/K+ ATPase enzyme system. Omeprazole, a proton pump inhibitor, binds covalently to cysteine residues on the alpha subunit of the H+/K+ ATPase pump via disulfide bridges, limiting stomach acid output for up to 36 hours. This antisecretory action is dose-dependent and inhibits both basal and induced acid secretion, regardless of the stimulus.
Metabolism
The cytochrome P450 (CYP) enzyme system metabolizes omeprazole extensively in the liver. The polymorphically expressed CYP2C19, which is responsible for the synthesis of hydroxyomeprazole, the predominant metabolite detected in plasma, is responsible for the majority of its metabolism. The rest is dependent on CYP3A4, which is responsible for the synthesis of omeprazole sulphone.

Half life
0.5-1 hour (for healthy subjects using a delayed-release capsule). 3 hours (due to hepatic impairment)
Toxicity
Overdoes are characterized by symptoms such as confusion, drowsiness, impaired vision, tachycardia, nausea, diaphoresis, flushing, headache, and dry mouth.
Structure

IUPAC Name
6-methoxy-2-[(4-methoxy-3,5-dimethylpyridin-2-yl)methylsulfinyl]-1H-benzimidazole
Lansoprazole
About the drug
Lansoprazole, sold under the brand name Prevacid, is a proton pump inhibitor (PPI) and a substituted benzimidazole. Lansoprazole was authorized by the United States Food and Drug Administration (FDA) on October 19, 1995.
Indications
Lansoprazole is a gastric acid secretion inhibitor that is approved for the short-term treatment of active gastric ulcers, active duodenal ulcers, erosive reflux oesophagitis, symptomatic gastroesophageal reflux disease, and NSAID-induced gastric and duodenal ulcers. It can be used to treat and maintain a variety of stomach diseases, including duodenal ulcers, NSAID-related gastric ulcers, and erosive esophagitis. Lansoprazole reduces the recurrence of stomach ulcers in patients with a documented history of gastric ulcers who also use NSAIDs on a regular basis. It is also beneficial in the treatment of hypersecretory diseases such as Zollinger-Ellison syndrome. Lansoprazole is effective against H. pylori when combined with amoxicillin and clarithromycin (triple therapy) or when given alone (dual therapy).
Mechanism of action
Lansoprazole is a prodrug that requires protonation in an acidic environment to become activated as a PPI. Lansoprazole, once protonated, can react with cysteine residues on parietal H+,K+-ATPase, notably Cys813 and Cys321, resulting in persistent disulfides. Because of their propensity to bind covalently to their targets, PPIs in general can provide persistent suppression of acid secretion.
Metabolism
Lansoprazole is mostly metabolized in the liver by the enzymes CYP3A4 and CYP2C19. The primary metabolites that occur are 5-hydroxy lansoprazole and the lansoprazole sulfone derivative.

Half life
Lansoprazole has a limited half life of about 2 hours or less. Because of its method of action, lansoprazole can effectively block acid secretion for 24 hours.
Toxicity
The most often reported adverse effects in lansoprazole-treated individuals compared to placebo include abdominal pain, constipation, diarrhea, and nausea. A case of toxic epidermal necrolysis (TEN), an uncommon but severe cutaneous reaction caused by lansoprazole, has been reported.
Structure
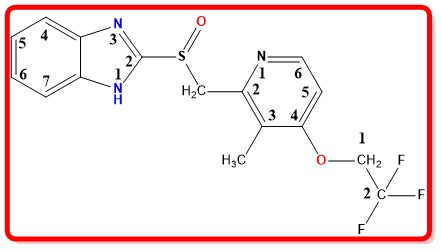
IUPAC Name
2-{[3-methyl-4-(2,2,2-trifluoroethoxy)pyridin-2-yl]methylsulfinyl}-1H-benzimidazole
Rabeprazole
About the drug
Rabeprazole is a proton pump inhibitor antiulcer medication. It is a prodrug that converts to active sulphenamide in the acid environment of the parietal cells. Rabeprazole, marketed as Aciphex, was approved by the United States Food and Drug Administration (FDA) on August 19, 1999.
Indications
NSAIDs are used to treat acid reflux disease (GERD), peptic ulcer disease, H. pylori eradication, and the prevention of gastrointestinal bleeding.
Mechanism of action
Rabeprazole belongs to a class of antisecretory compounds (substituted benzimidazole proton-pump inhibitors) that do not have anticholinergic or histamine H2-receptor antagonist properties but suppress gastric acid secretion by inhibiting the gastric H+/K+ATPase (hydrogen-potassium adenosine triphosphatase) at the secretory surface of the gastric parietal cell. Because this enzyme is thought to be the acid (proton) pump within the parietal cell, rabeprazole has been classified as a gastric proton-pump inhibitor. Rabeprazole inhibits the final stage of stomach acid output. Rabeprazole is protonated, accumulates, and is converted into an active sulfenamide in gastric parietal cells. Rabeprazole is chemically active at pH 1.2 and has a half-life of 78 seconds when tested in vitro.
Metabolism
Hepatic (Rabeprazole → Active metabolite of Rabeprazole)
Half life
1-2 hours
Structure

IUPAC Name
2-{[4-(3-methoxypropoxy)-3-methylpyridin-2-yl]methylsulfinyl}-1H-benzimidazole
Pantoprazole
About the drug
Pantoprazole is a proton pump inhibitor (PPI) of the first generation.
Mechanism of action
The secretion of hydrochloric acid (HCl) into the gastric lumen is primarily regulated by the proton pump’s H(+)/K(+)-ATPase, which is abundant in the stomach’s parietal cells. ATPase is an enzyme found on the parietal cell membrane that enables hydrogen and potassium exchange, which generally results in potassium extrusion and the creation of HCl (gastric acid).
Pantoprazole are substituted benzimidazole derivatives, weak bases that accumulate in the acidic area of the parietal cell before being transformed to active sulfenamide derivatives in the canaliculi (small canal) of the gastric parietal cell, also in an acidic environment. This active form then forms disulfide connections with critical cysteines on the gastric acid pump, preventing it from functioning.
Pantoprazole specifically binds to the sulfhydryl group of H+, K+-ATPase, an enzyme involved in speeding the final step in the acid secretion route. The enzyme is rendered inactive, preventing stomach acid secretion. Pantoprazole, inhibit stomach acid output more strongly and for a longer period of time than H(2) antagonists.
Metabolism
The cytochrome P450 (CYP) system metabolizes pantoprazole extensively in the liver. Pantoprazole metabolism is unaffected by administration route (intravenous or oral). The major metabolic process is demethylation by the hepatic cytochrome enzyme CYP2C19, followed by sulfation; other metabolic pathways include CYP3A4 oxidation.
Almost 80% of an oral or intravenous dose is eliminated as metabolites in urine after hepatic metabolism; the remaining is found in feces and is the result of biliary secretion.

Half life
About 1 hour
Toxicity
Pantoprazole was found to be carcinogenic in long-term rodent experiments, and its administration resulted in rare kinds of gastrointestinal cancers. At this moment, the relevance of these findings on tumor development in humans is unknown.
Structure

IUPAC Name
6-(difluoromethoxy)-2-[(3,4-dimethoxypyridin-2-yl)methylsulfinyl]-1H-benzimidazole
Therapeutic uses of proton pump inhibitors
Proton pump inhibitors (PPIs) are used to treat a variety of gastrointestinal disorders. PPIs are commonly used to treat the following conditions:
- PPIs are often used to treat gastroesophageal reflux disease (GERD), a disorder characterized by chronic acid reflux and heartburn. They aid to lower stomach acid production and alleviate discomfort.
- PPIs are used to treat peptic ulcers, which can form in the stomach (gastric ulcers) or the first part of the small intestine (duodenal ulcers). By lowering stomach acid levels, PPIs aid in the repair and prevention of ulcers.
- Infection with Helicobacter pylori: PPIs are frequently used in conjunction with antibiotics to treat Helicobacter pylori, a bacteria that can cause peptic ulcers and other gastrointestinal problems.
- PPIs are used to treat Zollinger-Ellison syndrome, a rare illness characterized by excessive stomach acid production. PPIs aid in the reduction of acid secretion and the relief of related symptoms.
- PPIs can be used to treat non-ulcer dyspepsia, a condition characterized by chronic or recurring pain or discomfort in the upper abdomen. PPIs alleviate symptoms by lowering stomach acid production.
- PPIs can be recommended to people who need long-term usage of nonsteroidal anti-inflammatory medicines (NSAIDs) to avoid the formation of ulcers in the stomach or duodenum caused by NSAIDs.
SAR of Proton pump inhibitors
General Structure

From the above general structure, following pattern can be draw so that SAR of proton pump inhibitor can be explained.

Aryl2
- The Aryl2 must be pyridine. Replacement of pyridine with other heterocyclic ring like pyrimidine or pyrazine leads to decrease in activity.
- Substitution on Aryl2 can be done on 3rd, 4th and 5th Among this 4th position substitution plays crucial role in activity.
- Presence of methoxy at 4th position increase the activity of compound. The methoxy group at position 4 of the pyridine ring gives electrons to the pyridine nitrogen. This raises not only the fraction of cationic pyridine but also the nucleophilic character of any proton pump ihibitor molecules with a unionized pyridine nitrogen. This enables intramolecular nucleophilic attack at C2 of the benzimidazole ring, resulting in the creation of active sulfenamide and sulfenic acid forms.
- Addition of methyl group on either 3rd position or 3rd and 5th position increase the activity. Positive induction, a sigma bond phenomenon, is used by the methyl groups at positions 3 and 5 to improve the nucleophilic nature of the unionized pyridine nitrogen.
Spacer
- Addition of one methyl group between linker and aryl2 portion leads to increase in activity.
- Increase or decrease in chain length will decrease the activity. The decrease in activity is due to increase in distance for intramolecular nucleophilic attack at C2 of the benzimidazole ring.
Linker
- Addition of [-C(O)-], [-C(N)-] or any other carba linkers leads to decrease in activity.
- Presence of [-S(O)-] as linker leads to increase in activity. Activity is increased to due formation of disulphide bonds. One thia of proton pump inhibitor and second thia of cysteine residue of proton pump involve in the disulphide bond formation.
Ary1
- To form active sulfenamide and sulfenic acid, intramolecular rearrangement is required. The Ary1 should be such that it should readily accept the electron from Ary2.
- 5-membered heterocyclic ring is suitable to fulfil this condition. The best rest is seen with imidazole.
- Addition of another aromatic homocyclic ring on imidazole nucleus produces benzimidazole.
- The benzimidazole’s pi electron donating action increases the electrophilicity of the C2 position.
- The 5-OCH3 group promotes N3 protonation, which draws electrons from C2 via sigma bonds.
- Although the OCH3 group has a negative inductive effect, the overall effect on the benzimidazole N3 is electronic enrichment.
Ask/Probable questions on this topic
MCQ questions
Previous year Ask questions
2 marks
……….
5 Marks
……….
10 marks
……….
MCQ
……….
Question Bank
2 marks
……….
5 Marks
……….
10 marks
……….
MCQ
……….
Smillingpharmacy prediction
……….