
BP607P – Medicinal Chemistry-III Lab Manual

Our Other Brands
Youtube channel

Telegram channel

Paid Advertisements

Free Courier service
Courier time: 8:00PM to 8:00 AM
Printing Charges
- 65 paisa per printing page for back to back printing – Market rate Rs 1.5 to Rs 10
- 75 paisa per xerox page for back to back xerox – Market rate Rs 1.5 per side
- Rs 4* colour printing/xerox – Market rate Rs 5 to Rs 10
- Rs 1.5 per printing page for back to back printing on bonded paper Market rate Rs 5 to Rs 8
- Rs 6* colour printing/xerox on bonded paper – Market rate Rs 7 to Rs 10
- A3 Print/Xerox black and white – Rs 10 – Market rate Rs 15 to Rs 25
- A3 colour Print – Rs 25 – Market rate Rs 40 to Rs 90
- Lamination A4 – Rs 15 – Market rate Rs 20 to Rs 25
- Spiral Binding-Rs 20 Market rate Rs 35 to Rs 90
Table of Contents
Syllabus as per - "PC and Autonomous"

- To study purification techniques of solvents/liquids and synthesized products by distillation, fractional distillation and distillation under vacuum and recrystallization.
- Demonstration of synthesis based on green chemistry (Microwave based)
- Preparation of TLC plates
- Determination of Melting point/range of the sample
- Drawing the structure of API by chembiodraw
- Determination of physicochemical properties by chembiodraw
- Conventional synthesis of sulfonamides (Step 1 – synthesis of acetanilide)
- Conventional synthesis of sulfonamides (Step 2 – synthesis of 4-acetamidobenzenesulphonyl chloride)
- Conventional synthesis of sulfonamides (Step 3 – synthesis of 4-acetamidoaniline)
- Conventional synthesis of sulfonamides (Step 4 – Final Step)
- Microwave synthesis of 1,3-pyrazole
- Microwave synthesis of 2,3-diphenyl quinoxaline
- Microwave synthesis of Benzocaine
- Microwave synthesis of phenytoin
- Microwave synthesis of methylsalicyclate
- Conventional synthesis of 7-hydroxy-4-methyl coumarin
Practical: 01
Blank page content
Aim
To study purification techniques of solvents/liquids and synthesized products by distillation, fractional distillation and distillation under vacuum and recrystallization.
Figures
Figure for Sublimation

Figure for distillation

Figure for fractional distillation

Figure for distillation under reduced pressure

Figure for steam distillation

Figure for differential extraction

Result
The purification techniques of solvents/liquids and synthesized products by various methods have been studied.
Line page content
Aim
To study purification techniques of solvents/liquids and synthesized products by distillation, fractional distillation and distillation under vacuum and recrystallization.
Reference
- Furniss B. S., Hannaford A.J., Smith P.W.G., Tatchell A.R., Vogel’s Textbook of Practical Organic Chemistry, Fifth Ed, Longman Sci and Technical, 1989, page 963.
- Ahluwalia V.K., Agrawal Renu., Organic Synthesis Special Techniques, Nerosa publishing house, 2001, p. no. 90 – 114
Requirements
Beakers, funnel, hot plate, filter paper pair of tongue, etc.
Theory
Purification in a chemical background is defined as the process of physical separation of a chemical substance of interest from foreign or contaminating substances. Pure compounds also called as isolate is a result of a successful purification process. There are different methods which can be applied individually or in combination to achieve isolate.
- Filtration is a mechanical method to separate solids from liquids or gases by passing the feed stream through a porous sheet such as a cloth or membrane, which retains the solids and allows the liquid to pass through.
- Centrifugation is a process for separation of tiny particles from the liquid which cannot be separated by filtration. The suspension is revolved at high speed with the help of an electric motor so that the fine particles get settle and liquid can be easily decanted.
- Evaporation is used to remove volatile liquids from non-volatile solutes.
- Liquid–liquid extraction removes an impurity or recovers a desired product by dissolving the crude material in a solvent in which other components of the feed material has less solubility.
- Crystallization separates a product from a liquid feed stream, often in extremely pure form, by cooling the feed stream or adding precipitants which lower the solubility of the desired product so that it forms crystals. The pure solid crystals are then separated from the remaining liquor by filtration or centrifugation. The difference in the solubility’s of an organic compound and that of the impurities present in it, in a suitable solvent is the basis for this method of separation. A concentrated solution is prepared by dissolving the compound in a suitable solvent; the pure compound crystallizes out on cooling. The dissolved impurities are removed through filtration. The filtrate contains some amount of pure compound too. Compound containing impurities of comparable solubility’s are purified by repeated crystallization.
- Recrystallisation: In analytical and synthetic chemistry work, purchased reagents of doubtful purity may be Recrystallize, e.g. dissolved in a very pure solvent, and then crystallized, and the crystals recovered, in order to improve and/or verify their purity.
- Distillation is a widely used in petroleum refining and in purification of ethanol separates volatile liquids on the basis of their relative volatilities. This method is mainly used for separating liquids with sufficient difference in their boiling points. It can also be used for separating volatile liquids from non-volatile impurities.
Purification of Organic compounds
Many methods viz., Simple and fractional crystallisation, sublimation, distillation (simple/fractional/reduced pressure/steam/ azetropic), chromatography, differential extraction and chemical methods are available for the purification of substances. The choice of method, however, depends upon the nature of substance (whether solid or liquid) and the type of impurities present in it.
Simple Crystallisation
This is the most common method used to purify organic solids. It is based upon the fact that whenever a crystal is formed, it tends to leave out the impurities. For crystallization, choice of suitable solvent depends on
- Solvent that dissolves more of the substance at higher temperature than at room temperature
- In which impurities are either insoluble or dissolve to an extent that they remain in solution (in the mother liquor) upon crystallization,
- Solvent which is not highly inflammable
- And which does not react chemically with the compound to be crystallized.
The most commonly used solvents for crystallization are: water, alcohol, ether, chloroform, carbon- tetrachloride, acetone, benzene, petroleum ether etc.
Examples:
- Sugar having an impurity of common salt can be crystallized from hot ethanol since sugar dissolves in hot ethanol but common salt does not.
- A mixture of benzoic acid and naphthalene can be separated from hot water in which benzoic acid dissolves but naphthalene does not.
Fractional Crystallization
The process of separation of different components of a mixture by repeated crystallisations is called fractional crystallisation. The mixture is dissolved in a solvent in which the two components have different solubility’s. When a hot saturated solution of this mixture is allowed to cool, the less soluble component crystallises out first while the more soluble substance remains in solution. The mother liquor left after crystallisation of the less soluble component is again concentrated and then allowed to cool when the crystals of the more soluble component are obtained. The two components thus separated are recrystallized from the same or different solvent to yield both the components of the mixture in pure form.
Fractional crystallisation can be used to separate a mixture of KCl and BaCl2, KCl (less soluble) and BaCl2 (more soluble).
Sublimation
Certain organic solids on heating directly change from solid to vapour state without passing through a liquid state, such substances are called sublimable and this process is called sublimation.
The sublimation process is used for the separation of sublimable volatile compounds from non sublimable impurities. The process is generally used for the purification of camphor, naphthalene, anthracene, benzoic acid, Iodine and salicylic acid etc containing non-volatile impurities.
Distillation
Distillation is the joint process of vaporisation and condensation. This method is used for the purification of liquids which boil without decomposition and contain non-volatile impurities. This method can also be used for separating liquids having sufficient difference in their boiling points. This method can be used to separate a mixture of
- Chloroform (b. p. 334 K) and Aniline (b. p. 457 K)
- Ether (b. p. 308 K) and Toluene (b. p. 384 K)
Fractional Distillation
This process is used to separate a mixture of two or more miscible liquids which have boiling points close to each other. Since in this process, the distillate is collected in fractions under different temperatures, it is known as fractional distillation. This process is carried out by using fractionating columns. Fractionating column is a special type of long glass tube provided with obstructions to the passage of the vapour upwards and that of liquid downwards. This method may be used to separate a mixture of acetone (b. p. 330 K) and methyl alcohol (b. p. 338 K) or a mixture of benzene and toluene. One of the technological applications of fractional distillation is to separate different fractions of crude oil in petroleum industry into various useful fractions such as gasoline, kerosene oil, diesel oil, lubricating oil etc.
Distillation under Reduced Pressure
This method is used for the purification of high boiling liquids and liquids which decompose at or below their boiling points. The crude liquid is heated in distillation flask fitted with a water condenser, receiver and vacuum pump. As the pressure is reduced, the liquid begins to boil at a much lower temperature than its normal boiling point. The vapour is condensed by water condenser and the pure liquid collects in the receiver.
Glycerol which decomposes at its boiling point (563 K) under atmospheric pressure can be distilled without decomposition at 453 K under 12 mm of Hg. Similarly, sugarcane juice is concentrated in sugar industry by evaporation under reduced pressure which saves a lot of fuel.
Steam Distillation
This method is applicable for the separation and purification of those organic compounds (solids or liquids) which (a) are insoluble in water (b) are volatile in steam (c) possess a high vapour pressure (10-15 mm Hg) at 373 K and (d) contain non-volatile impurities.
Aniline (b. p. 457 K) can be purified by steam distillation since it boils at a temperature of 371.5 K in presence of steam. Other compounds which can be purified by steam distillation are: nitrobenzene, bromobenzene, o-nitrophenol, salicylaldehyde, o-hydroxyacetophenone, essential oils, turpentine oil etc.
Azeotropic Distillation
Azeotropic mixture is a mixture having constant boiling point. The most familiar example is a mixture of ethanol and water in the ratio of 95.87: 4.13 (a ratio present in rectified spirit). It boils at 78.13oC. The constituents of an azeotropic mixture can’t be separated by fractional distillation. Hence a special type of distillation (azeotropic distillation) is used for separating the constituents of an azeotropic mixture.
In this method a third compound is used in distillation. The process is based on the fact that dehydrating agents like diethyl ether etc. depress the partial pressure of one of the original components. As a result, the boiling point of that component is raised sufficiently and thus the other component will distil over.
Dehydrating agents having low boiling point (e.g. ether) depress the partial pressure of alcohol more than that of water; on the other hand, dehydrating agents having high boiling point (glycerol, glycol) depress the partial pressure of water more than that of alcohol.
Chromatography
This is a modern method used for the separation of mixtures into its components, purification of compounds and also to test the purity of compounds. The name chromatography is based on the Greek word ‘chroma’ meaning colour and ‘graphy’ for writing because the method was first used for the separation of coloured substances found in plants. This method was described by Tswett in 1906.
Principle of chromatography:
The technique of chromatography is based on the difference in the rates at which the components of a mixture move through a porous medium (called stationary phase) under the influence of some solvent or gas (called moving phase). Thus, this technique consists of two phases- one is a stationary phase of large surface area while the second is a moving phase which is allowed to move slowly over the stationary phase. The stationary phase is either a solid or a liquid while the moving phase may be a liquid or a gas.
Types of chromatography
Depending upon the nature of the stationary and the mobile phases, the different types of chromatographic techniques commonly used are in each table
Type of Chromatography | Mobile/Stationary Phase | Uses |
Adsorption or column chromatography | Liquid/Solid | Large scale separations |
Thin-layer chromatography | Liquid/Solid | Qualitative analysis (Identification and characterization of organic compounds) |
High performance liquid chromatography | Liquid/Solid | Qualitative and quantitative analysis |
Gas-liquid chromatography (GLC) | Gas/Liquid | Qualitative and quantitative analysis |
Partition chromatography or ascending paper chromatography | Liquid/Liquid | Qualitative and quantitative analysis of polar organic compounds (sugars, a-amino acids and inorganic compounds) |
Differential Extraction
This method is used for the separation of an organic compound (solid or liquid) from its aqueous solution by shaking with a suitable solvent (e.g. ether, benzene, chloroform, carbon tetrachloride etc.) in a separating funnel. The selected solvent should be immiscible with water but should dissolve the organic compound to an appreciable extent. It is important to note that extraction is more efficient (i.e., more complete) when a given volume of the extracting solvent is used in several installments. This method is normally applied to non-volatile compounds. For example, benzoic acid can be extracted from its water solution using benzene.
Chemical Methods
Besides these physical methods, a number of chemical methods have also been used to separate a mixture of organic compounds. These methods are based upon the distinguishing chemical properties of one class of organic compounds from the others. Phenols can be separated from carboxylic acids on treatment with an aqueous solution of sodium bicarbonate. Since carboxylic acids dissolve in solution evolving but phenols usually do not react. Destructive distillation of wood gives pyroligneous acid which contains acetic acid (10%), acetone (0.5%) and methanol (3%). Acetic acid can be separated from this mixture by treating it with milk of lime when acetic acid forms the calcium salt. The reaction mixture on distillation gives a mixture of acetone and methanol (which can be further separated by fractional distillation into individual components as mentioned above) while the calcium salt remains as residue in the flask. The calcium salt is then decomposed with dil HCl and distilled to afford acetic acid. A mixture of 1o, 2o and 3o amines can be separated using either benzenesulphonyl chloride (Hinsberg’s reagent) or diethyl oxalate (Hoffmann’s method).
Criteria of purity of organic compounds
The purity of an organic compound can be ascertained by determining its some physical constants like m.p., b.p., specific gravity, refractive index and viscosity. In usual practice, sharp m.p. (in case of solids) and boiling point (in case of liquids) are used as criteria for purity because their determination is feasible in the laboratory. A pure organic solid has a definite and sharp (sudden, rapid and complete) melting point, while an impure substance has a lower and indefinite melting point.
Mixed melting point
The melting point of two thoroughly mixed substances is called mixed melting point. This can also be used for ascertaining the purity of a compound.
The substance, whose purity is to be tested, is mixed with a pure sample of the same compound. The melting point of the mixture is determined. If the melting point of the mixture is sharp and comes out to be the same as that of pure compound, it is sure that the compound under test is pure. On the other hand, if the melting point of the mixture is less than the melting point of the pure compound, the compound in question is not pure.
Result
The purification techniques of solvents/liquids and synthesized products by various methods have been studied.
Practical No 2
Blank page content
Aim
Demonstration of synthesis based on green chemistry (Microwave based)
Figures
Figure of Principle

Figure of Dielectric

Figure of microwave process

Figure of Conventional versus microwave process

Result
The demonstration to microwave assisted green synthesis was studied carefully
Line page content
Aim
Demonstration of synthesis based on green chemistry (Microwave based)
References
Ahluwalia V.K., Agrawal Renu., Organic Synthesis: Special Techniques, Narosa Book Distributors (2009).
Theory
Introduction
The bottleneck of conventional synthesis is typically the optimization, i.e. finding the optimum conditions for a specific reaction to obtain the desired products in good yields and purities. Since many synthesis reactions require at least one or more heating steps for long time periods, these optimizations are often difficult and time-consuming. Microwave-assisted heating under controlled conditions has been shown to be a valuable technology for any application that requires heating of a reaction mixture, since it often dramatically reduces reaction times – typically from days or hours to minutes or even seconds. Compounds can therefore be rapidly synthesized in either a parallel or (automated) sequential way using this new promising technology.
Principle
Microwave irradiation is electromagnetic irradiation in the frequency range 0.3 to 300 GHz, corresponding to wavelengths between 1 mm to 1 m. All domestic “kitchen” microwave ovens as well as commercially available dedicated microwave reactors for chemical synthesis operate at a frequency of 2.45 GHz.
As can be precisely calculated, the energy of microwave irradiation is too low to cleave molecular bonds. It is therefore clear that microwaves cannot “induce” chemical reactions by direct absorption of microwave power. However, microwave irradiation provides unique thermal effects, which are highly beneficial for chemical synthesis. Cleaving molecular bonds and therefore inducing chemical reactions is only possible employing irradiation with higher energy
Microwave dielectric heating
Microwave chemistry is based on the efficient heating of materials (in most cases solvents) by dielectric heating effects. Dielectric heating works by two major mechanisms
Dipolar polarization
For a substance to be able to generate heat when irradiated with microwaves it must be a dipole, i.e. its molecular structure must be partly negatively and partly positively charged. Since the microwave field is oscillating, the dipoles in the field align to the oscillating field. This alignment causes rotation, which results in friction and ultimately in heat energy.
Ionic conduction
During ionic conduction, dissolved (completely) charged particles (usually ions) oscillate back and forth under the influence of microwave irradiation. This oscillation causes collisions of the charged particles with neighboring molecules or atoms, which are ultimately responsible for creating heat energy. As an example: if equal amounts of distilled water and tap water are heated by microwave irradiation, more rapid heating will occur for the tap water because of its ionic content in addition to the dipolar rotation of water molecules.
Dielectric properties
As the term “dielectric heating” suggests, a material must possess certain dielectric properties in order to be efficiently heated in the microwave field. The heating characteristics of a particular material (e.g. a solvent) under microwave irradiation conditions are dependent on the ability of a specific substance to convert electromagnetic energy into heat. This ability is determined by the so-called loss tangent, tan δ.

Where,
ε′′ = dielectric loss = efficiency with which electromagnetic radiation is converted into heat.
ε′ = dielectric constant = polarizibility of molecules in the electric field
The tan δ values for some commonly used organic solvents are summarized in Table given below.
This table shows the classification of solvents into high (tan δ > 0.5), medium (tan δ 0.1-0.5), and low microwave absorbing (tan δ < 0.1). Solvents without a dipole moment, such as benzene and dioxane, are more or less microwave transparent (tan δ < 0.01).
A solvent with a high tan δ is required for rapid heating in the microwave field. However, this does not mean that solvents with low tan δ values cannot be used for microwave synthesis. Since either substrates or reagents/catalysts are likely to be polar, the overall dielectric properties of a reaction mixture will in most cases allow sufficient heating by microwaves, even with non-polar solvents. If, however, the mixture is non-polar, passive heating elements can be added to aid the heating process.
High (> 0.5) | Medium (0.1 – 0.5) | Low (< 0.1) | |||
Solvent | tan δ | Solvent | tan δ | Solvent | tan δ |
Ethylene glycol | 1.350 | 2-Butanol | 0.447 | Chloroform | 0.091 |
Ethanol | 0.941 | Dichlorobenzene | 0.280 | Acetonitrile | 0.062 |
DMSO | 0.825 | NMP | 0.275 | Ethyl acetate | 0.059 |
2-Propanol | 0.799 | Acetic acid | 0.174 | Acetone | 0.054 |
Formic acid | 0.722 | DMF | 0.161 | THF | 0.047 |
Methanol | 0.659 | Dichloroethane | 0.127 | Dichloromethane | 0.042 |
Nitrobenzene | 0.589 | Water | 0.123 | Toluene | 0.040 |
1-Butanol | 0.571 | Chlorobenzene | 0.101 | Hexane | 0.020 |
In general, the interaction of microwave irradiation with matter is characterized by three different processes: absorption, transmission and reflection. While highly dielectric materials, like polar organic solvents, lead to strong absorption of microwaves and consequently to a rapid heating of the medium, non-polar (microwave transparent) materials show only small interactions with microwaves (transmission). Microwaves pass through such materials. This makes them suitable as construction materials for reactors. If microwave radiation is reflected by the material surface, there is almost no introduction of energy into the system.
Microwave vs. conventional heating
Traditionally, organic synthesis is carried out by refluxing a reaction mixture using a hot oil bath as a heat source. However, this way of heating a reaction mixture is comparatively slow and energy inefficient, since first the heat energy is transferred from the hot oil bath to the surface of the reaction vessel, and then the hot surface heats the content of the reaction vessel. Furthermore, the hot surface can lead to local overheating and to decomposition of sensitive material.
In contrast, microwave irradiation results in energy efficient internal heating by direct coupling of microwave energy with dipoles and/or ions that are present in the reaction mixture. Microwaves pass through the microwave-transparent vessel wall and heat the reaction mixture on a molecular basis – by direct interaction with the molecules (solvents, reagents, catalysts, etc.). Due to this direct “in-core” heating (no initial heating of the vessel surface), microwave irradiation results in inverted temperature gradients as compared to a conventionally heated system.
Furthermore, the conversion of electromagnetic energy into heat energy works highly efficiently and results in extremely fast heating rates – not reproducible with conventional heating. Due to the rapid heating to the target temperature, the formation of by-products is suppressed. This is another huge advantage of microwave heating, since it means that higher product yields can be achieved and the work-up is simplified.
Microwave-Processing Techniques
- Solvent free reactions
- Phase transfer catalyst
- Reactions using solvents
- Parallel processing
Common reactions occur in microwave synthesis
- Cycloaddition reactions Diels–Alder cycloaddition
- Asymmetric Allylic Alkylations
- Glycosylation Reaction
- Nucleophilic Aromatic Substitution
- Oxidations
- Buchwald-Hartwig Reaction
- Ullmann Condensation Reactions
Benefits of microwave synthesis in dedicated microwave reactors
- Possibility of convenient superheating of solvents
- Excellent parameter control
- Access to automated setups and parallel synthesis
- Possibility of stirring
- Continuous power output
- Intuitive user interface via touchscreen
- Utmost safety even under high temperature/pressure conditions
Demerits of Microwave Synthesis
- In microwave synthesis sudden increase in temperature may led to the distortion of molecules which may lead to distortion of the reaction.
- Reactions are very vigorous, and which may be hazardous.
- Microwave reactors are expensive and very delectated so there must be a care to be taken during their use.
- Short reaction period, so a care must be taken during the process.
- Various reactions which have short reaction time are not be undertaken in microwave reactor due to sudden increase in the temperature it may be hazardous, and it may lead to reaction crises.
- Many other things like, temperature sensitive reactions, reactions involving bumping of material, and reaction in which effervescences and colour reaction are not be done in Microwave reactor.
Result
The demonstration to microwave assisted green synthesis was studied carefully
Practical 3
Blank page content
Aim
Demonstration of reaction monitoring by TLC
Working model of TLC

Observations
- Distance travelled by starting material: ______ cm
- Distance travelled by synthesized compound: ______ cm
- Distance travelled by solvent: ______ cm
- Rf value of starting material: ______ cm
- Rf value of synthesized compound: ______ cm
Calculations

Result
The Rf value of starting material and synthesized compound are _____ and _____ which clears that the product was formed/not formed and have/does not have any starting material or other impurities.
Line page Content
Aim
Demonstration of reaction monitoring by TLC
References
Jeffery G. H., Basset J., Mendhan J., Denny R. C. Vogel’s text book of qualitative chemical analysis, fifth edition, longmanscientific and technical publishing house, 1989, page no
Requirement
Chemical
Mobile phase, organic solvents, silica gel G, Samples
Apparatus
Glass Chamber, Glass plate
Instruments
Iodine chamber/UV Cabinet
Theory
TLC is based on adsorption type of chromatography. The solute molecules get adsorbed by electrostatic force on silica gel. Difference in electrostatic forces of solutes molecules leads to their separation. The separation of solute is also based on the type of mobile phase use.
TLC is an extremely valuable analytical technique in the organic lab. It provides a rapid separation of compounds, and thereby gives an indication of the number and nature of the components of a mixture. TLC can also be used to identify compounds by comparison with known samples, to check the purity of a compound, or to monitor the progress of a reaction, an extraction, or a purification procedure.
Principle
TLC is normally done on a small glass or plastic plate coated with a thin layer of a solid. The most common are silica (SiO2) or alumina (Al2O3). This is the stationary phase. The mobile phase is an organic solvent or solvent mixture. The sample mixture is applied near the bottom of the plate as a small spot, and then placed in a jar containing a few ml of solvent. The solvent climbs up the plate by capillary action, carrying the sample mixture along with it. Each compound in the mixture moves at a different rate, depending on its solubility in the mobile phase and the strength of its absorption to the stationary phase. When the solvent gets near the top of the plate, it is allowed to evaporate, leaving behind the components of the mixture at various distances from the point of origin. The ratio of the distance a compound moves to the distance the solvent moves is the Rf value (retention factor). This value is characteristic of the compound, the solvent, and the stationary phase.
Procedure
Preparation of TLC Plate
Before glass plates are coated with adsorbent, they must be carefully cleaned with laboratory detergent, using a test-tube brush to remove adhering particles, rinsed thoroughly with distilled water, placed in a suitable metal rack and dried in an oven. After the treatment with detergent solution the plates should only be handled by the edges or by the under-surface which is not to be coated with adsorbent. Failure to observe this precaution may result in the formation of a mechanically unstable layer which is liable to flaking due to grease spots on the glass surface. In severe cases of grease contamination, it may be necessary to use a chromic acid cleaning mixture. Small plates are conveniently prepared from microscope slides using a dipping technique; this operation is conducted in a fume cupboard. Slurry is prepared by the slow addition with shaking of 30g of adsorbent to 100ml of dry dichloromethane contained in a wide-necked capped bottle. A pair of microscope slides is held together and dipped into the slurry, slowly withdrawn and allowed to drain momentarily while held over the bottle. The slides are parted carefully and placed horizontally in a rack sited in a fume cupboard to dry for approximately 10 minutes. The surplus adsorbent is then removed by means of a razor blade drawn down the glass edges as shown in the figure.
It may be desirable to activate the adsorbent further by heating it at 110°C.
Development of Chromatogram

- Take the glass chamber; add the prepared mobile phase. Add the filter paper for efficient saturation of the developing chamber.
- Take the activated glass plate to it and apply the starting material with the help of micropipette. Allow the starting material solution to dry.
- With the help of a second micropipette, apply synthesized product solution on the same activated plate at another location. Allow the synthesized product solution to dry.
- Put this plate in the developing chamber slowly without flushing.
- Allow the solvent front to run 80% of plate height.
- After achieving the desired target of solvent front, remove the plate and allow drying for 5 minutes.
- Put the dry plate in an iodine chamber or spray the suitable reagent to locate the solute spot.
- Calculate the Rf value of starting material and synthesized compound. Put the values on the observation table.
Result
The Rf value of starting material and synthesized compound are _____ and _____ which clears that the product was formed/not formed and have/does not have any starting material or other impurities.
Practical 4
Blank page content
Aim
Determination of Melting point of the sample
Figures
Sealing of capillary

Filling of capillary

Tying of capillary tube to thermometer

Determination of Melting point

Observation table
Sr. No | Name of sample | Start Point | End point | Melting point/Range |
1 |
|
|
|
|
2 |
|
|
|
|
3 |
|
|
|
|
4 |
|
|
|
|
Result
The melting point/range of the given sample was found to be –
1.
2.
3.
4.
Line page content
Aim
Determination of Melting point of the sample
Reference
Brain S. Furniss; Antony J. Hannford; Peter W. G. smith; Austin R. Tatchell. Vogel’s Textbook of practical organic chemistry, Pearson Education, New Delhi, 5th edition, page No –
Requirements
Apparatus
Theil’s tube, capillary tube, thermometer, thread, magnifying glass, black paper, match box
Chemical
Liquid praffin, dry sample powder
Theory
The change from solid to liquid state of a compound in heating is called melting and the temperature at which a solid in its pure form melts is called the melting point. Every pure solid has a characteristics melting point therefore determination of melting point helps in identification of the compound. Presence of impurities lowers the melting point of the solid. Thus, Melting point also serves as a criterion of purity of a compound.
A pure crystalline organic compound has, in general has definite and sharp melting point; that is, the melting point range (the difference between the temperature at which the collapse of the crystals is first observed and the temperature at which the sample becomes completely liquid) does not exceed about 0.5 °C. The presence of small quantities of miscible, or partially miscible, impurities will usually produce a marked increase in the melting point range and cause the commencement of melting to occur at a temperature lower than the melting point of the pure substance. In this case, melting point named as melting range. The melting point is therefore a valuable criterion of purity for an organic compound.
Procedure
The experimental method in most common use is to heat a small amount (about 1 mg) of the substance in a capillary tube inserted into a suitable melting point apparatus and to determine the temperature at which melting occurs. The capillary melting point tubes are prepared either from soft glass test tubes or from wide glass tubing.
Sealing of capillary tube
One end of each of the capillary tubes should be sealed by inserting it horizontally into the extreme edge of a small Bunsen flame for a few seconds, and the capillary tube rotated meanwhile; the formation of a glass bead at the end of the tube should be avoided.
Filling the capillary tube
The capillary tube is then filled as follows. About 25 mg of the dry substance is placed on a glass slide or upon a fragment of clean, porous porcelain plate and finely powdered with a clean metal or glass spatula, and then formed into a small mound. The open end of the capillary tube is pushed into the powder, ‘backing’ the latter, if necessary, with a spatula. The solid is then shaken down the tube by tapping the closed end on the bench or by gently drawing the flat side of a triangular file (a pocket nail file is quite effective) along the upper end of the tube. The procedure is repeated until the length of lightly packed material is 3-5 mm, and the outside of the tube is finally wiped clean.
Another method is rubbing the capillary tube on the rough surface. Due to vibrations, the sample settle at bottom. Both methods take time for travelling solid sample towards sealed end. The most time saving unreported method is use of condenser. The powder was tapped at the open end of capillary tube. This filled capillary tube is allowed to drop in between the condenser. Pickup the capillary tube, you will see that that solid sample has reached to sealed end.
Tying the capillary tube to thermometer
The filled melting point tube is attached to the lower end of the thermometer in such a way that the substance is at the level of the middle of the mercury bulb (which has previously been wetted with the bath liquid); the moistened capillary is then slid into position. The thermometer, with capillary attached, is inserted into the centre of the main tube of the Thiele apparatus.
Determination of melting point/range
The safest and most satisfactory bath liquids are the highly stable and heat-resistant Silicone oils. A cheaper alternative is medicinal paraffin; it has a low specific heat, is non-flammable and is non-corrosive, but it can only be safely heated to about 220°C; above this temperature it begins to decompose and becomes discoloured.
On heating the bent side-arm, the heated liquid circulates and raises the temperature of the sample in such a way that no stirring of the bath liquid is required. The melting point apparatus is heated comparatively rapidly with a small flame until the temperature of the bath is within 15°C of the melting point of the substance, and then slowly and regularly at the rate of about 2°C per minute until the compound melts completely. The temperature at which the substance commences to liquefy and the temperature at which the solid has disappeared, i.e. the melting point range, is observed. For a pure compound, the melting point range should not exceed.0.5-1 °C; it is usually less.
Result
The melting point/range of the given sample was found to be –
1.
2.
3.
4.
Practical No 5
Blank page content
Aim
Drawing Structures of Active Pharmaceutical Agents Using ChemBioDraw Software
Figures
Software basic window
Toolbar for selecting ring system
Toolbar for drawing bonds

Toolbar for editing item
Result
Successfully drawn the structures of selected active pharmaceutical agents using ChemBioDraw software.
Line page content
Aim
Drawing Structures of Active Pharmaceutical Agents Using ChemBioDraw Software
Reference
- User Guide of ChemBioDraw Software.
- Textbook of Pharmaceutical Chemistry by Beale and Block.
- Research papers on the applications of ChemBioDraw in drug design and development
Material required
- Computer
- ChemBioDraw Software (installed on a computer)
- List of active pharmaceutical agents.
- Laboratory notebook or digital documentation tool
Theory
ChemBioDraw is a powerful chemical structure drawing software widely used in pharmaceutical and chemical research. It helps in visualizing molecular structures, designing chemical reactions, and predicting molecular properties. The software provides tools for drawing accurate 2D representations of molecules, which are crucial in pharmaceutical drug development and documentation.
Active pharmaceutical agents (APAs) are the biologically active components in drug formulations responsible for therapeutic effects.
Applications of ChemBioDraw in Pharmaceuticals
- Drawing and annotating chemical structures.
- Predicting properties like logP, molecular weight, and more.
- Generating IUPAC names for molecules.
- Designing reaction schemes.
Procedure
- Launching ChemBioDraw:
- Open ChemBioDraw on the computer.
- Familiarize yourself with the toolbar and workspace.
- Drawing Structures:
- Select a chemical structure to draw (e.g., Paracetamol).
- Use tools from the toolbar to:
- Add carbon chains by clicking and dragging.
- Introduce functional groups (e.g., hydroxyl, amine) using the functional group selector.
- Adjust bond angles and lengths to make the structure precise.
- Annotating the Structure:
- Label atoms where necessary.
- Add stereochemical indicators (e.g., R/S, cis/trans).
- Analyzing the Structure:
- Use the “Analysis” tools to calculate molecular properties (e.g., molecular weight, polar surface area).
- Generate the IUPAC name of the structure.
- Saving and Exporting:
- Save the file in ChemBioDraw format (.cdx) for future edits.
- Export the structure as an image (.png, .jpeg) for documentation or presentations.
- Repeat for Other Molecules:
- Follow the same steps for additional active pharmaceutical agents.
Result
Successfully drawn the structures of selected active pharmaceutical agents using ChemBioDraw software.
Practical No 6
Blank page content
Aim
To calculate the molecular weight, logP, molar refractivity, pKa and elemental analysis values for following drugs by chemdraw professional Software
Observation
Sr. No | Name of API | Structure | Mol. Wt. | Log P | MR | pKa | Elemental Analysis |
1 |
|
|
|
|
|
| C: H: Cl: F: N: O: S: |
2 |
|
|
|
|
|
| C: H: Cl: F: N: O: S: |
3 |
|
|
|
|
|
| C: H: Cl: F: N: O: S: |
4 |
|
|
|
|
|
| C: H: Cl: F: N: O: S: |
5 |
|
|
|
|
|
| C: H: Cl: F: N: O: S: |
Result
The molecular weight, logP, molar refractivity, pKa and elemental analysis values of selected 5 molecules were successfully calculated.
Line page content
Aim
To calculate the molecular weight, logP, molar refractivity, pKa and elemental analysis values for following drugs by chemdraw professional Software
Theory
Theory
Molecular weight
The molecular weight is the mass of one mole of a substance. The molecular weight of compound is calculated by adding the atomic mass of each atom present in the molecule.
Molar refractivity
Molar refractivity is a measure of the total polarizability of a mole of substance that depends on temperature, index of refraction and pressure.
The molar refractivity is defined as
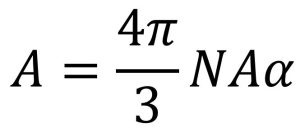
LogP value
LogP Value is used to determine the solubility of compound and is defined as concentration of drug in organic layer to concentration of drug in aqueous layer.
pKa value
pKa is the negative logarithm of the acid dissociation constant (Ka) and is used to express the strength of an acid in solution. It is a fundamental property in medicinal chemistry that helps predict the ionization state of a compound at a given pH. pKa is a critical parameter for understanding drug behavior in biological systems and plays a vital role in pharmaceutical development, aiding in the design of effective and safe medications.
Elemental Analysis
Elemental analysis is a technique used to determine the quantitative composition of elements (such as carbon, hydrogen, nitrogen, oxygen, sulfur, and others) in a compound. It is an essential tool in medicinal chemistry to verify the molecular formula of synthesized compounds, ensure purity, and assess the presence of any unexpected elements or impurities.
Procedure
- Draw the structure in chem biodraw
- Check for any error
- Click on Analysis tools provided in View option
- Click on chemical properties that is also provided in view option
- Transfer the calculated data in the table.
Result
The molecular weight, logP, molar refractivity, pKa and elemental analysis values of selected 5 molecules were successfully calculated.
Practical No 7
Blank page content
Aim
Determination of Molecular Properties, Lipinski’s Rule of Five, and Bioactivity Score of Selected API
Result
The molecular properties, Lipinski’s Rule compliance, and bioactivity scores of the selected APIs were successfully determined and analyzed.
Line page content
Aim
Determination of Molecular Properties, Lipinski’s Rule of Five, and Bioactivity Score of Selected API
Reference
- Lipinski, C. A. et al. “Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings.” Advanced Drug Delivery Reviews (2001).
- Molinspiration Chemoinformatics: molinspiration.com
- SwissADME: swissadme.ch
- Drug-Likeness in Medicinal Chemistry, Concepts and Applications, Wiley.
Requirement
- Computational tools or online platforms (e.g., Molinspiration, SwissADME, or ChemDraw software)
- Selected APIs (e.g., Paracetamol, Ibuprofen, Aspirin)
- Laboratory notebook for documentation
Theory
Molecular Properties: Molecular properties such as molecular weight, logP (partition coefficient), hydrogen bond donors (HBD), and hydrogen bond acceptors (HBA) are crucial in understanding the pharmacokinetic behavior of a drug. These properties influence solubility, permeability, and distribution of drugs in the body.
Lipinski’s Rule of Five: Proposed by Christopher Lipinski, the “Rule of Five” predicts the oral bioavailability of drugs. According to this rule, a compound is more likely to be orally active if:
Procedure
Input the Chemical Structure:
- Draw the structure of the selected API using ChemDraw or any molecular drawing tool.
- Save the structure in SMILES format or any compatible file format for computational tools.
Determine Molecular Properties:
- Open the computational tool (e.g., SwissADME or Molinspiration).
- Upload the chemical structure or input the SMILES notation.
- Analyze the molecular properties such as molecular weight, logP, HBD, and HBA.
Assess Lipinski’s Rule of Five:
- Compare the calculated molecular properties against Lipinski’s criteria.
- Note any violations of the rule.
Calculate Bioactivity Score:
- Use a bioactivity prediction tool (e.g., Molinspiration).
- Input the molecular structure.
- Record the bioactivity scores for different biological targets.
Interpret Results:
- Determine if the compound adheres to Lipinski’s Rule of Five.
- Assess the likelihood of bioactivity based on the scores.
Result
The molecular properties, Lipinski’s Rule compliance, and bioactivity scores of the selected APIs were successfully determined and analyzed.
Practical No 8
Blank page content
Aim
Microwave assist synthesis of 3-methyl-1-phenyl pyrazol-5-one from ethyl acetoacetate
Observation
- Weight of watch glass + phenyl hydrazine :_________g
- Weight of watch glass after transfer : ________g
- Weight of phenyl hydrazine taken (1-2) (SAY A) :_________g
- Molecular weight of phenyl hydrazine :_________ g
- Molecular weight of 3-methyl-1-phenyl pyrazol-5-one :_________ g
- Weight of container :_________ g
- Weight of Container + 3-methyl-1-phenyl pyrazol-5-one :_________ g
- Weight of synthesized 3-methyl-1-phenyl pyrazol-5-one :_________ g
Reaction

Reaction Mechanism

Calculation

Result

Line page content
Aim
Microwave assist synthesis of 3-methyl-1-phenyl pyrazol-5-one from ethyl acetoacetate
Reference
Sharma S. V., Badami S., Venkateshwarlu L., Suresh B. Microwave technology in pharmaceutical chemistry practical: Synthesis of organic drugs. Indian Drugs, 40(8), 2003, page no 452
Requirement
Apparatus
Beaker, stirrer, measuring cylinder, thermometer, thiel’s tube
Chemical
Ethyl acetoacetate, phenylhydrazine, ether
Instrument
Theory
The Knorr pyrazole synthesis is an organic reaction used to convert a hydrazine or its derivatives and a 1,3-dicarbonyl compound to a pyrazole using an acid catalyst. The mechanism begins with an acid catalysed imine formation, where in the case of hydrazine derivatives the attack can happen on either carbonyl carbon or result in two possible products. The other nitrogen of the hydrazine derivative then attacks the other carbonyl group which has also been protonated by the acid and forms a second imine group. This diimine compound gets deprotonated to regenerate the acid catalyst and provide the final pyrazole product.
Procedure
- In a dry beaker, take ___g (1g) phenyl hydrazine
- To it add ___ml (1ml) of ethyl acetoacetate.
- Transfer the beaker to microwave
- Irradiate the content at 80% intensity for 6 minutes, 30 seconds.
- A reddish-brown syrupy liquid was obtained after cooling.
- Add ___ml (20ml) diethyl ether was added to it with vigorous stirring.
- A yellow colour compound slowly separated.
- Filter the product and at pump and wash it with ether to removed coloured impurities.
- Dry the product and determine the practical yield, melting point and check the purity of compound by performing TLC.
- Input all the analytical values in the table
Result
Practical No 9
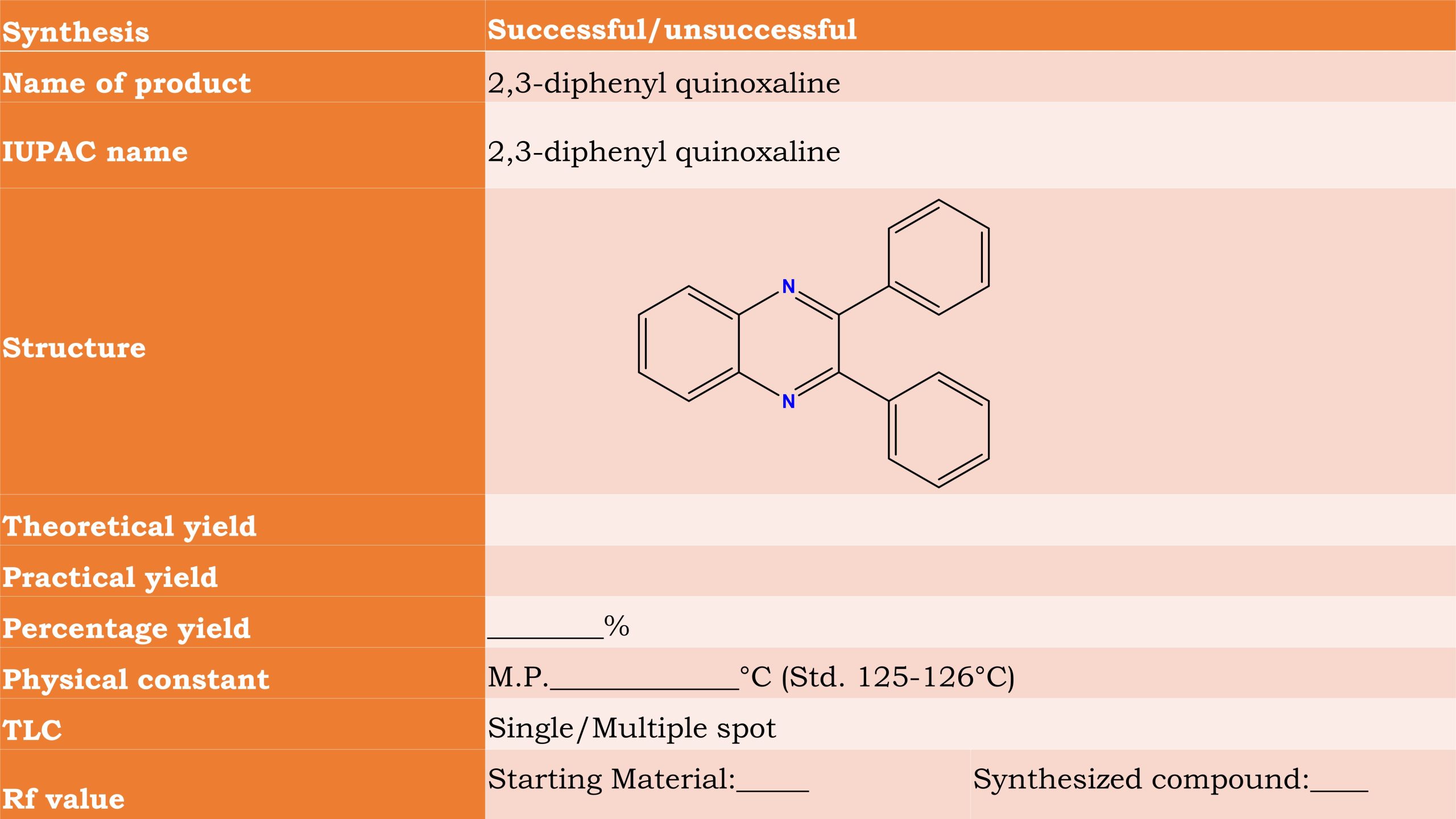
Microwave synthesis of 2,3-diphenyl quinoxaline
Observation
- Weight of watch glass + Benzil : __________g
- Weight of watch glass after transfer : __________g
- Weight of Benzil taken (1-2) (SAY A) : __________g
- Molecular weight of Benzil :_________ g
- Molecular weight of 2,3-diphenyl quinoxaline :_________ g
- Weight of container :_________ g
- Weight of Container + 2,3-diphenyl quinoxaline :_________ g
- Weight of synthesized 2,3-diphenyl quinoxaline :_________ g
Reaction

Reaction Mechanism

Calculation

Result
Line page content
Aim
Microwave synthesis of 2,3-diphenyl quinoxaline
Reference
Sharma S. V., Badami S., Venkateshwarlu L., Suresh B. Microwave technology in pharmaceutical chemistry practical: Synthesis of organic drugs. Indian Drugs, 40(8), 2003, page no 452
Requirement
Apparatus
Beaker, Stirrer, measuring cylinder, water bath
Chemical
o-diphenylene diamine, ethanol, benzil
Instrument
Microwave, Balance, vacuum pump
Theory
This synthesis is based on condensation reaction between benzil and o-phenylene diamine
Procedure
- In a dry moltar and pestle take ___g (1g) of
- To it add ____g (0.5g) of o-phenylene diamine
- Transfer the solid content in a beaker.
- Transfer the beaker to microwave
- Irradiate the solid at 100% intensity for 90 seconds.
- The brown liquid was obtained in hot conditions which after cooling get solidify.
- Dissolve the solid in methanol and add a pinch of activated charcoal
- Boil the solution on water bath and filter in hot condition
- Keep the clear filtrate in ice bath.
- Crystal of obtained
- Add ____ml (8ml) of rectified spirit
- Collect the solid by vacuum filtration and wash with ice-cold water.
- Determine the practical yield, melting point and check the purity of compound by performing TLC.
- After completion of analytical evaluation, pack the compound, label it and submit it.
- Input all the analytical values in the table
Result
Practical No 10
Blank page content
Aim
Microwave synthesis of Benzocaine
Observation
- Weight of watch glass + p-amino benzoic acid :_________ g
- Weight of watch glass after transfer :_________ g
- Weight of p-amino benzoic acid taken for synthesis :_________ g (Say A)
- Molecular weight of p-amino benzoic acid :_________ g
- Molecular weight of Benzocaine :_________ g
- Weight of container :_________ g
- Weight of Container + Benzocaine :_________ g
- Weight of synthesized Benzocaine :_________ g
Reaction

Reaction Mechanism

Calculation

Result

Line page content
Aim
Microwave synthesis of Benzocaine
Reference
Sharma S. V., Badami S. Microwave technology in pharmaceutical chemistry practical: Synthesis of organic drugs. 40(08), 2003, page No 451
Requirement
Apparatus
Conical flask, funnel, beaker, microscopic slide
Chemical
p-amino benzoic acid, ethanol, Concentrated H2SO4, Sodium carbonate, silica gel G
Instrument
Microwave instrument, vaccum filtration assembly, Weighing balance
Theory
The synthesis of benzocaine is based on the esterification reaction. The acid part of benzoic acid undergoes condensation with ethanol to form benzocaine
Procedure
- In a clean dry flask, Add ___g (0.5g) of p-amino benzoic acid
- To it add place ____ml (6ml) of dry ethanol and mix the content.
- Add drop wise, ____ml (1ml) of concentrated sulphuric acid and mix the content.
- Transfer the flask to microwave. Cover the flask by funnel.
- A beaker containing water was keep near the reaction vessel to provide sink condition.
- Irradiate the solution with 20% intensity for 6 minutes.
- The pale brown colour liquid was obtained. It was cooled in water.
- Add the 10% sodium carbonate solution till the pH 9 is achieved.
- The white precipitate is obtained.
- Filter the precipitate through suction.
- Dry the compound in air
- Determine the practical yield, melting point and check the purity of compound by performing TLC.
- After completion of analytical evaluation, pack the compound, label it and submit it.
- Input all the analytical values in the table
Result
Practical No 11
Blank page content
Aim
Microwave synthesis of Phenytoin
Observation
- Weight of watch glass + Benzil :_________ g
- Weight of watch glass after transfer :_________ g
- Weight of Benzil taken for synthesis :_________ g (Say A)
- Molecular weight of Benzil :_________ g
- Molecular weight of phenytoin :_________ g
- Weight of container :_________ g
- Weight of Container + phenytoin :_________ g
- Weight of synthesized phenytoin :_________ g
Reaction

Reaction Mechanism

Calculation

Result

Line page content
Aim
Microwave synthesis of Phenytoin
Reference
Mukesh K. Nagar*, Karishma R. Waghmare2, P.N. Dhabale, Pradeep D. Chanekar and Suresh Bhatia. Microwave Assisted Synthesis and Characterization of Phenytoin. Asian J. Research Chem. 4(4): April, 2011; Page 619-620.
Requirement
Apparatus
RBF, Condenser, Beaker, Measuring cylinder, Water bath, Funnel
Chemical
Benzil, Sodium hydroxide, Concentrated HCl, Urea, Water
Instrument
Microwave Suction filtration assembly, IR Lamp, Weighing balance
Theory
The synthesis of benzil is based on 1,2 shift type of reaction resembles to benzillic acid. Sodium hydroxide acts as a catalyst. Ethanol is used to dissolve benzil.
Procedure
- In a clean dry conical flask, take ____g (0.53g) of Benzil.
- To it add ____g (0.3g) of urea.
- Add ____ml (3ml) of 30% aqueous sodium hydroxide solution.
- To it add ___ml (3ml) of methanol. Put the funnel on the conical flask.
- Irradiate the solution at 40% intensity for 4 minutes.
- Mixture starts boiling after 30 seconds and white solid get separated.
- After 4 minutes, cool the content and pour the reaction mixture in to ____ml (30ml) water and mix.
- Allow to stand for 15 minutes and filter the precipitate to remove insoluble debris.
- Acidify the filtrate with Conc. HCl. Cool in ice and filter immediately via suction.
- Determine the product yield, % yield, melting point and TLC and report the date in the table
Result